




Зірабев як представник найчисельнішої «родини» біосимілярів у галузі онкології
Резюме. На біологічні препарати припадає велика частка «фармацевтичних» витрат у галузі онкології — близько половини, за деякими оцінками [1]. Щоб задовольнити наявні потреби та підвищити доступність нових методів лікування, розробляють наступні версії біологічних ліків, які можуть потрапляти на ринок після закінчення патентного захисту оригінальних. Порівняно з референтними препаратами біосиміляри, як правило, дешевші та доступніші; отже, вони мають потенціал для значного зниження витрат на сферу охорони здоров’я. Бевацизумаб, моноклональне антитіло, є першим лікарським засобом, який ефективно використовує молекулярний сигнальний шлях фактора росту судинного ендотелію (vascular endothelial growth factor — VEGF) і його рецептор (VEGF-R) як фармакологічну мішень. Великий клінічний потенціал цього препарату є підґрунтям для масштабної роботи із досліджень та розширення сфери його застосування у Європейському Союзі (ЄС) та США [2]. Свідченням успішного запровадження результатів досліджень є кількість схвалених біосимілярів бевацизумабу — 9, найбільша у ЄС серед ліків, що застосовують у галузі онкології [3]. При цьому позиція європейського регулятора полягає в тому, що після схвалення біосиміляри є взаємозамінними з їхнім оригінальним препаратом і між собою, із екстраполяцією усіх показань до медичного застосування без додаткових клінічних досліджень [4, 5]. Такий підхід не означає зниження планки регуляторних вимог, адже у його основі — особливий акцент на аналітичній частині досьє, що потребує акумуляції значних зусиль та досвіду від заявників. Нерідко ними є авторитетні інноваційні фармацевтичні компанії, такі як Pfizer, яка у 2019 р. отримала дозвіл на маркетинг у ЄС та США препарату бевацизумабу Зірабев / Zirabev® із екстраполяцією усіх показань референтного лікарського засобу Авастин / Avastin.
DOI: 10.32471/clinicaloncology.2663-466X.55-3.33091
 Біосиміляр, за визначенням ЕМА, — це «біологічний лікарський засіб, що містить версію діючої речовини вже затвердженого оригінального біологічного препарату (референтного)» [6]. Біосиміляри мають значну схожість з референтними препаратами за складом, профілем безпеки, ефективністю та імуногенністю, але не ідентичні їм. Біосиміляри та оригінальні біологічні препарати в ЄС оцінюються за схожими вимогами та стандартами. Це забезпечує відсутність клінічно важливих відмінностей щодо чистоти, безпеки та ефективності [1].
Біосиміляр, за визначенням ЕМА, — це «біологічний лікарський засіб, що містить версію діючої речовини вже затвердженого оригінального біологічного препарату (референтного)» [6]. Біосиміляри мають значну схожість з референтними препаратами за складом, профілем безпеки, ефективністю та імуногенністю, але не ідентичні їм. Біосиміляри та оригінальні біологічні препарати в ЄС оцінюються за схожими вимогами та стандартами. Це забезпечує відсутність клінічно важливих відмінностей щодо чистоти, безпеки та ефективності [1].
Регуляторний шлях схвалення біоподібних лікарських засобів на рівні ЄС вперше розроблено у 2005 р., що дало змогу Європейському агентству з лікарських засобів (European Medicines Agency — EMA) дозволити маркетинг першого біоподібного препарату Omnitrope (соматотропін) у 2006 р. [6]. У США регуляторну процедуру започатковано завдяки Закону про конкуренцію цін на біологічні препарати та інновації (Biologics Price Competition and Innovation Act — BPCIA) у 2010 р., що дозволило Управлінню з контролю за харчовими продуктами та лікарськими засобами (Food and Drug Administration — FDA) створити настанову для розробників [7] та схвалити перший біосиміляр Zarxio (філграстим-sndz) у 2015 р.
Починаючи із 2020 р., коли FDA започаткувало «Фіолетову книгу» (Purple Book), — онлайн-базу даних про схвалені FDA біологічні продукти, необхідну релевантну інформацію про референтні біологічні препарати та біосиміляри можна знайти в цьому джерелі [8].
Регуляторні вимоги до біосимілярів продовжують розвиватися. Так, у 2015 р. в ЄС набула чинності переглянута тематична настанова [9], а у 2018 р. в США розроблено «План дій FDA щодо біоподібних препаратів» (Biosimilars Action Plan) [10].
Хоча щодо схвалення біосимілярів на основі встановлення біоподібності з референтним препаратом існує глобальний консенсус, між підходами 2 провідних регуляторів є деякі відмінності: EMA надає рекомендації для конкретних груп продуктів, тоді як FDA, яке розробило свої рекомендації набагато пізніше, використовує особливий підхід до кожного препарату [7, 11].
Також існує концептуальна різниця у значенні взаємозамінності, яке використовується в США, порівняно з рештою світу. У США взаємозамінність — це особливий статус біоподібного препарату, який надається FDA після виконання додаткових вимог, таких як клінічні дослідження зі зміною режимів лікування між групами досліджуваних (multi-switch trial) [3]. Навпаки, EMA у 2023 р. опублікувала позицію про взаємозамінність біосимілярів з їхнім референтним лікарським засобом і між собою [4, 12].
Однак обидві регуляторні агенції мають подібні вимоги до заявників щодо надання даних про загальну біоподібність, що передбачає поетапний підхід до порівнянь, починаючи із великого пакета аналітичних даних (рисунок) [11].
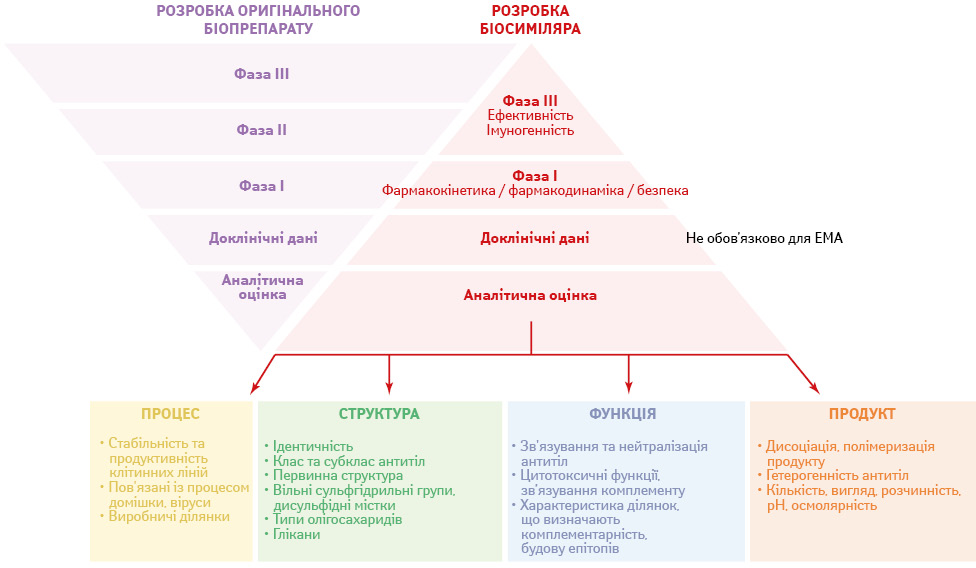
Крім того, для забезпечення ефективності та безпеки в довгостроковій перспективі важливі дані фармаконагляду [5].
Біосиміляри в галузі онкології
Наразі закінчується термін дії численних патентів на біологічні препарати, і кількість схвалюваних біосимілярів стрімко збільшується. У ЄС дозвіл на маркетинг за централізованою процедурою станом на 28.09.2024 р. отримали 123 таких лікарських засоби [13], у США — 59, з яких 17 мають статус взаємозамінних [8, 14]. Близько 1/3 МНН схвалених біосимілярів належить до тих, що застосовують у сфері онкології, зокрема антинеопластичних засобів (включно з 3 —моноклональними антитілами) (таблиця).
| Міжнародне непатентоване найменування (МНН) із зазначенням в АТС-класифікації |
ЕМА | FDA (зокрема взаємозамінних) |
| L01F. Антинеопластичні засоби. Моноклональні антитіла | ||
| Бевацизумаб (інгібітор VEGF / VEGF-R) | 9 | 5 (0) |
| Ритуксимаб (інгібітор CD20 (кластерів диференціювання 20)) | 5 | 3 (0) |
| Трастузумаб (інгібітор HER2 (рецептора епідермального фактора росту людини 2)) | 7 | 6 (0) |
| В03Х. Інші антианемічні препарати | ||
| Еритропоетин | 5 | 1 (0) |
| L03AA. Імуностимулятори. Колонієстимулюючі фактори | ||
| Філграстим | 7 | 4 (0) |
| Пегфілграстим | 8 | 7 (0) |
Всесвітня організація охорони здоров’я (ВООЗ) із програмою прекваліфікації [16], авторитетні професійні асоціації, такі як Американське товариство клінічної онкології (American Society of Clinical Oncology — ASCO), Європейське товариство медичної онкології (European Society for Medical Oncology — ESMO) та Національна мережа багатопрофільних онкологічних закладів США (National Comprehensive Cancer Network — NCCN) розробляють керівні принципи та виступають із освітніми ініціативами, сприяючи підвищенню практичної користі біосимілярів [17–19]. Зокрема, біосиміляри, схвалені FDA, такі як бевацизумаб-bvzr, рекомендовані як варіанти лікування в Клінічних рекомендаціях NCCN [19].
Також збільшується доказова база щодо взаємозамінності біосимілярів бевацизумабу, ритуксимабу та трастузумабу, що може ще більше підвищити довіру лікарів та пацієнтів [3]. Наприклад, дослідники з Нідерландів, проаналізувавши публічні звіти з оцінки реєстраційного досьє (European Public Assessment Report — EPAR) усіх схвалених у ЄС біосимілярів бевацизумабу, дійшли висновку про надійне документальне підтвердження усіх клінічно релевантних атрибутів [3].
Можливості екстраполяції
Екстраполяцією називають науково-регуляторний процес затвердження показань до медичного застосування біоаналогів, спираючись на аналогічний такому у референтного препарату механізм дії, не потребуючи власних даних про ефективність [9]. Такий підхід дозволяє заощадити кошти розробників, оскільки необхідно проводити менш масштабні клінічні дослідження.
Препарат Зірабев
Бевацизумаб — це рекомбінантне гуманізоване моноклональне антитіло, яке зв’язується з фактором росту ендотелію судин і блокує його, тим самим інгібуючи ангіогенез і ріст пухлин [20]. У 2004 р. FDA схвалило бевацизумаб для використання як препарат першої лінії в лікуванні метастатичного колоректального раку поєднано з хімієтерапією на основі фторурацилу. Пізніше, у 2019 р., дозвіл на маркетинг у США та ЄС отримав біосиміляр Зірабев (Pfizer) [3]. Показання до призначення біосиміляра аналогічні референтному препарату та включають лікування у дорослих пацієнтів [20]:
- метастатичного колоректального раку у дорослих пацієнтів у комбінації з хімієтерапією на основі похідних фторпіримідину;
- метастатичного раку молочної залози у дорослих пацієнтів у комбінації з паклітакселом чи капецитабіном;
- нерезектабельного поширеного метастатичного чи рецидивного недрібноклітинного раку легень у комбінації з хімієтерапією на основі похідних платини;
- нерезектабельного поширеного метастатичного чи рецидивного неплоскоклітинного недрібноклітинного раку легень з EGFR-активуючими мутаціями (EGFR — рецептор епідермального фактора росту) у комбінації з ерлотинібом;
- поширеного та/або метастатичного нирково-клітинного раку в комбінації з інтерфероном альфа-2а;
- поширеного та рецидивного епітеліального раку яєчників, фаллопієвої труби і первинного раку очеревини у дорослих хворих у комбінації з карбоплатином і паклітакселом чи іншими препаратами;
- персистуючого, рецидивного або метастатичного раку шийки матки в комбінації з паклітакселом і цисплатином або в якості альтернативи з паклітакселом і топотеканом.
З чого складається клінічна доказова база препарату Зірабев (досліджуваного лікарського засобу PF-06439535)? У даних з ClinicalTrials.gov можна виявити декілька фармакокінетичних досліджень (I фази за участю здорових добровольців), дослідження переносу (bridging study) із вивчення ефективності, безпеки, фармакокінетики та імуногенності бевацизумабу у пацієнтів монголоїдної та негроїдної рас та проспективне відкрите багатоцентрове постмаркетингове вивчення ефективності та безпеки [21]. Для бевацизумабу такі дослідження проводять за участю хворих з недрібноклітинним раком легень (НДКРЛ) із використанням кінцевих точок, достатньо чутливих, щоб визначити будь-які суттєві клінічні відмінності між біологічним і біоподібним продуктом [3].
Так, у подвійному сліпому дослідженні N. Reinmuth та співавт. (2019) пацієнтів із 159 центрів у 27 країнах рандомізовано у співвідношенні 1:1, у групи отримання PF-06439535 (біосиміляра бевацизумабу) та референтного бевацизумабу; у обох — поєднано з паклітакселом і карбоплатином у 1-шу добу кожного 21-денного циклу протягом 4–6 циклів [22]. Результати підсумкового аналізу даних після завершення дослідження свідчать, що помітних відмінностей у показниках виживаності без прогресування (ВБП) або загальній виживаності (ЗВ) між групами не зафіксовано. Найчастішими побічними ефектами (ПЕ) ≥III ступеня, що виникли під час лікування, були розвиток артеріальної гіпертензії, нейтропенії та анемії. При цьому не встановлено клінічно значущих відмінностей у безпеці, фармакокінетиці чи імуногенності між групами. Так, серед пацієнтів із поширеним неплоскоклітинним НДКРЛ ефективність біосиміляра бевацизумабу PF-06439535 була схожою з еталонним бевацизумабом з ЄС та США при порівнювальному профілі безпеки [22, 23].
Також у рамках постмаркетингових клінічних досліджень вивчено застосування біосимілярів бевацизумабу, зокрема Зірабеву, при дуже широкому спектрі онкологічних захворювань. Слід зазначити, що ініціаторами так званих досліджень перепрофілювання здебільшого виступають клініки та наукові центри [23]. У таких клінічних дослідженнях часто одночасно застосовують біосиміляри від різних виробників. Взаємозамінність цих препаратів, з точки зору європейського регулятора, підвищує потенціал їх спільного застосування.
Відповідальність виробника щодо відстеження результатів застосування біосиміляра в реальній клінічній практиці також реалізується завдяки дослідницьким зусиллям. Так, починається залучення у проспективне відкрите неінтервенційне багатоцентрове постмаркетингове спостереження для оцінки безпеки та ефективності Зірабеву (біоподібного бевацизумабу) у пацієнтів з недрібноклітинним раком легень, метастатичним колоректальним раком, метастатичними новоутвореннями молочної залози, поширеною або метастатичною пухлиною нирки, раком шийки матки, епітеліальним новоутворенням яєчників, раком фаллопієвої труби, первинною перитонеальною онкопатологією або мультиформною гліобластомою (NCT04889495). Сплановане для оцінки кількох найважливіших параметрів ефективності та безпеки, воно надасть інформацію про користь препарату в умовах реальної клінічної практики.
Отже, Зірабев має дуже широку сферу застосування в клінічній онкології. Він також реалізує свій потенціал щодо отримання нових показань до застосування.
список Використаної літератури
1. Konstantinidou, S., Papaspiliou, A., & Kokkotou, E. (2020). Jan Current and future roles of biosimilars in oncology practice. Oncol. Lett.,19(1), 45–51. doi: 10.3892/ol.2019.11105.
2. Bachu, R. D., Abou-Dahech, M., & Tiwari, A. K. (2022). Oncology biosimilars: New developments and future directions. Cancer Rep. (Hoboken), 5(11), e1720. doi: 10.1002/cnr2.1720.
3. Broer, L. N., Knapen, D. G., & Lub-de Hooge, M. N. (2024). Monoclonal antibody biosimilars for cancer treatment. iScience, 27(6), 110115. doi: 10.1016/j.isci.2024.110115.
4. Heads of Medicines Agencies. European Medicines Agency (2023). Statement on the scientific rationale supporting interchangeability of biosimilar medicines in the EU, EMA / 627319 / 2022. Retrieved from http://www.ema.europa.eu/ en/documents/ public-statement/statement- scientific-rationale- supporting- interchangeability-biosimilar-medicines-eu_en.pdf.
5. Nabhan, C., Parsad, S., & Feinberg, B. A. (2018). Biosimilars in oncology in the United States: a review. JAMA Oncology, 4(2), 241–247. doi: 10.1001/jamaoncol.2017.4034.
6. European Medicines Agency (2005). Guideline on similar biological medicinal products. CHMP/437/04. Retrieved from http://www.ema.europa.eu/en/similar-biological-medicinal-products-scientific-guideline.
7. US Food and Drug Administration (2015). Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Retrieved from http://www.fda.gov/media/82647/download.
8. FDA. Purple Book. Database of Licensed Biological Products, purplebooksearch.fda.gov. Accessed 26.09.2024.
9. European Medicines Agency (2015). Guideline on similar biological medicinal products (Rev. 1). CHMP/437/04. Accessed 26.09.2024, Retrieved from
http://www.ema.europa.eu/en/similar-biological-medicinal-products-scientific-guideline.
10. FDA (2018). Biosimilars Action Plan: Balancing Innovation and Competition; Retrieved from http://www.fda.gov/media/114574/download.
11. Daller, J. (2016). Biosimilars: A consideration of the regulations in the United States and European Union. Regul. Toxicol. Pharm., 76, 199–208. doi: 10.1016/j.yrtph.2015.12.013.
12. Gherghescu, I., & Delgado-Charro, M. B. (2020). The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA. Pharmaceutics, 13(1), 48. doi: 10.3390/pharmaceutics13010048.
13. EMA. Medicines for Human Use. Accessed 26.09.2024. Retrieved from http://www.ema.europa.eu/en/medicines.
14. FDA-Approved Biosimilar Products. Accessed 26.09.2024. Retrieved from http://www.fda.gov/ drugs/ biosimilars/ biosimilar-product- information.
15. MHRA (2022). Guidance on the licensing of biosimilar products. Retrieved from http://www.gov.uk/government/ publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products.
16. Medicines (Finished Pharmaceutical Products / Biotherapeutic products) — Prequalification. Accessed 26.09.2024. Retrieved from extranet.who.int/prequal/ medicines/prequalified/finished-pharmaceutical-products.
17. Rodriguez, G., Mancuso, J., Lyman, G. H., Cardoso, F., Nahleh, Z., Vose, J. M., … Sherwood, S. (2023). ASCO Policy Statement on Biosimilar and Interchangeable Products in Oncology. JCO Oncol. Pract.,19(7), 411–419. doi: 10.1200/OP.22.00783.
18. Schiestl, M., & Krendyukov, A. (2017) The ESMO position paper on biosimilars in oncology: enhancing the provision of accurate education and information. ESMO Open., 2(3), e000245. doi: 10.1136/esmoopen-2017-000245.
19. NCCN (2021). NCCN Pharmacy Directors Forum Recommendations on Operationalizing the Safe and Efficient Use of Biosimilars in the Clinical Setting. Retrieved from http://www.nccn.org/ docs/default-source/ clinical/nccn-pharmacy- directors-forum-white- paper-operationalizing- the-safe-and- efficient-use- of-biosimilars.pdf?sfvrsn=136b3110_6.
20. ЗІРАБЕВ, інструкція для медичного застосування (РП № UA/18148/01/01).
21. U.S. Department of Health and Human Services, National Institutes of Health, National Library of Medicine, and National Center for Biotechnology Information. A Post-marketing Surveillance to Assess the Safety and Effectiveness of Zirabev in Domestic Patients With Various Cancer. ClinicalTrials.gov. ID NCT 04889495
22. Reinmuth, N., Bryl, M., Bondarenko, I., Syrigos, K., Vladimirov, V., Zereu, M., … Kasahara, K. (2019). PF-06439535 (a Bevacizumab Biosimilar) Compared with Reference Bevacizumab (Avastin®), Both Plus Paclitaxel and Carboplatin, as First-Line Treatment for Advanced Non-Squamous Non-Small-Cell Lung Cancer: A Randomized, Double-Blind Study. BioDrugs, 33(5), 555–570. doi: 10.1007/s40259-019-00363-4.
23. Ivanov, A., Getova-Kolarova, V., & Getov, I. (2024). Research and pilot analysis of bevacizumab repurposing potential and its impact in clinical practice. Biotechnology & Biotechnological Equipment, 38(1). doi: 10.1080/13102818.2024.2357191.
Ірина Неміш, Дар’я Полякова





Leave a comment