




Первинні Т-клітинні лімфоми шкіри: класифікація, діагностика, лікування (огляд літератури)
Крячок І.А., Алексик О.М., Титоренко І.Б.
Резюме. Останніми роками відмічено великий прогрес щодо поліпшення виживаності хворих на лімфоми. Одним із найбільш актуальних питань є діагностика та лікування Т-клітинних лімфом шкіри з використанням у діагностичній панелі широкого спектра сучасних методів обстеження (гістологічних, імунологічних, молекулярно-генетичних), які дають змогу правильно визначити прогноз та оптимізувати терапію із застосуванням індивідуалізованих підходів до лікування за допомогою новітніх методів.
Одержано 27.07.2023
Прийнято до друку 9.08.2023
DOI: 10.32471/clinicaloncology.2663-466X.50-2.30797
Первинні Т-клітинні шкірні лімфоми є гетерогенною групою лімфом, що характеризується клональною проліферацією Т-лімфоцитів у шкірі. Вони становлять 75–80% всіх шкірних лімфом. Первинні лімфоми шкіри відрізняються від нодальних лімфом за характером перебігу, прогнозом та підходами до терапії. Ці особливості викликали необхідність створення докладної класифікації, що відображатиме увесь спектр первинних шкірних лімфом (табл. 1). У 2018 р. на основі класифікації ВООЗ для пухлин гемопоетичної та лімфоїдної тканин та класифікації лімфом шкіри Європейської організації з дослідження та лікування раку (European Organisation for. Research and Treatment of Cancer — EORTC) створено ВООЗ/EORTC-класифікацію шкірних лімфом [1]. Сучасна класифікація лімфом комплексна і враховує інформацію про їх морфологію, імунофенотип, генетичні та клінічні особливості. У 2022 р. було анонсовано нове — 5-те видання класифікації гематологічних лімфоїдних пухлин ВООЗ [2].
| WHO-EORTC класифікація первинних лімфом шкіри, 2018 | Міжнародна консенсусна класифікація(ICC) зрілих лімфоїдних новоутворень (2022) | Класифікація ВООЗ гематолімфоїдних пухлин: лімфоїдні новоутворення (5-те видання, 2022) |
| Синдром Сезарі | Синдром Сезарі | Синдром Сезарі |
| Первинні Т-клітинні лімфоми шкіри | ||
| Грибоподібний мікоз | Грибоподібний мікоз | Грибоподібний мікоз |
Варіанти грибоподібного мікозу
|
Не включено | Не включено |
Лімфопроліферативні захворювання:
|
Лімфопроліферативні захворювання:
|
Лімфопроліферативні захворювання:
|
| Підшкірна панікулітоподібна Т-клітинна лімфома | Підшкірна панікулітоподібна Т-клітинна лімфома | Підшкірна панікулітоподібна Т-клітинна лімфома |
| Первинна шкірна периферична Т-клітинна лімфома, підтипи, які відмічають рідко
Первинний шкірний CD4+ малий або середній Т-клітинний лімфопроліферативний розлад (тимчасово) Первинна шкірна гамма/дельта Т-клітинна лімфома Первинна шкірна акральна CD8+ Т-клітинна лімфома Первинна шкірна агресивна епідермотропна CD8+ цитотоксична Т-клітинна лімфома (тимчасово) Первинна шкірна периферична Т-клітинна лімфома, NOS |
||
| Первинний шкірний CD4+ малий або середній Т-клітинний лімфопроліферативний розлад | Первинний шкірний CD4+ малий або середній Т-клітинний лімфопроліферативний розлад | |
| Первинна шкірна гамма/дельта Т-клітинна лімфома | Первинна шкірна гамма/дельта Т-клітинна лімфома | |
| Первинний шкірний акральний CD8+ лімфопроліферативний розлад | Первинний шкірний акральний CD8+ лімфопроліферативний розлад | |
| Первинна шкірна CD8+ агресивна епідермотропна цитотоксична Т-клітинна лімфома | Первинна шкірна CD8+ агресивна епідермотропна цитотоксична Т-клітинна лімфома | |
| Не включено | Первинна шкірна периферична Т-клітинна лімфома, NOS |
Шкірні Т-клітинні лімфоми відрізняються за перебігом. До індолентних Т-неходжкінських лімфом (НХЛ) належать грибоподібний мікоз, первинні шкірні CD30-позитивні лімфопроліферативні захворювання, підшкірна панікулітоподібна Т-клітинна лімфома, CD4+ дрібно-/середньоклітинна плеоморфна шкірна Т-клітинна лімфома (ШТКЛ), до агресивних — синдром Сезарі (СС), CD8+ агресивна епідермотропна цитотоксична ШТКЛ, шкірна Y/δ Т-клітинна лімфома.
Причини та патогенез Т-клітинних лімфом шкіри до кінця не з’ясовані. Нині більшість дослідників розглядають вірус Т-клітинного лейкозу людини 1-го типу (HTLV-1) як основний етіологічний фактор, що ініціює розвиток Т-клітинних злоякісних лімфом шкіри. Поряд із цим у розвитку Т-клітинної лімфоми обговорюється роль інших вірусів: Епштейна — Барр, герпесу 6-го типу. У хворих на Т-клітинну лімфому віруси виявляють у шкірі, периферичній крові, клітинах Лангерганса. Антитіла до HTVL-I виявляють у багатьох хворих на грибоподібний мікоз.
Клінічні спостереження вказують на можливу трансформацію тривалих хронічних дерматозів (нейродерміт, атопічний дерматит, псоріаз та ін.) в грибоподібний мікоз. При цьому ключовим фактором є тривала персистенція лімфоцитів у вогнищі запалення, які порушують імунний нагляд та зумовлюють появу клону злоякісних лімфоцитів та, таким чином, розвитку злоякісного проліферативного процесу.
Вплив на організм фізичних факторів, таких як інсоляція, іонізуюча радіація, хімічні речовини, може призвести до появи клону «генотравматичних» лімфоцитів, що виявляють мутагенний ефект на лімфоїдні клітини та сприяють малігнізації лімфоцитів.
Найпоширенішими підтипами Т-клітинних НХЛ є грибоподібний мікоз, синдром Сезарі, первинна анапластична великоклітинна лімфома шкіри та лімфоматоїдний папульоз. Вони становлять близько 95% всіх Т-клітинних шкірних лімфом.
Грибоподібний мікоз — первинна епідермотропна Т-клітинна лімфома шкіри, відмінною рисою якої є проліферація Т-лімфоцитів малих та середніх розмірів із церебриформними ядрами. Це найчастіша Т-клітинна пухлина шкіри, що становить 1% всіх НХЛ і 50% Т-клітинних шкірних лімфом. Захворюваність у світі становить 0,36–0,9 випадку на 100 000 населення [3, 4]. Середній вік хворих — 55–60 років, співвідношення чоловіків та жінок — 2:1 [5].
Класична форма грибоподібного мікозу характеризується трьома стадіями розвитку: еритематозно-сквамозною, бляшковою та пухлинною.
Гістологічна картина на ранніх стадіях грибоподібного мікозу неспецифічна і може бути схожою на подібну при доброякісному запальному дерматозі: відзначаються периваскулярні інфільтрати в поєднанні з псоріазоформною гіперплазією епідермісу [6]. Для бляшкової стадії характерний щільний смугоподібний інфільтрат у верхній частині дерми, що містить велику частку церебриформних лімфоцитів із вираженим епідермотропізмом. Внутрішньоепідермальні скупчення атипових лімфоцитів (мікроабсцеси Потріє) є характерною особливістю цієї стадії, але відмічаються лише в 10% випадків [7]. З прогресуванням у пухлинну стадію епідермотропізм зникає, а інфільтрат, що складається з церебриформних лімфоцитів малих, середніх та великих розмірів, стає дифузним і може проникати в підшкірну жирову клітковину [8].
Пухлинні клітини при грибоподібному мікозі мають фенотип зрілих Т-лімфоцитів пам’яті (CD3+, CD4+, CD45RO+, CD8-). Рідко можуть виявляти фенотип CD4-, CD8+. При грибоподібному мікозі часто відмічається втрата пан-Т-клітинних антигенів CD2, CD3, CD5, CD7, що у багатьох випадках є важливим доповненням до діагнозу [9].
Клінічна картина при грибоподібному мікозі дуже різноманітна, відзначається висип варіабельної форми, розмірів і кольору, який часто супроводжується свербежем, також характерні множинність висипу, кілька зон залучення, локалізація висипу на ділянках шкіри, що не піддаються сонячному опроміненню, пойкілодермія. Для грибоподібного мікозу характерний феномен одночасного прогресування та регресування окремого висипу.
Стадіювання грибоподібного мікозу проводиться згідно з рекомендаціями Міжнародного товариства з лімфом шкіри та Європейської організації з вивчення та лікування раку для грибоподібного мікозу та синдрому Сезарі (СС) (ISCL-EORTC staging system for MF/SS) (табл. 2) [10].
| Т (шкіра) | ||||
| Т1 | Обмежена пляма/бляшка (ураження <10% загальної поверхні шкіри) | |||
| Т2 | Генералізовані плями/бляшки (ураження ≥10% загальної поверхні шкіри) | |||
| Т3 | Пухлина(і) | |||
| Т4 | Еритродермія | |||
| N (лімфатичний вузол) | ||||
| N0 | Без клінічно збільшених периферичних лімфатичних вузлів | |||
| N1 | Клінічно збільшені периферичні лімфатичні вузли, що гістологічно не уражені | |||
| N2 | Клінічно збільшені лімфатичні вузли, гістологічно уражені (незмінена структура лімфатичного вузла) | |||
| N3 | Клінічно збільшені лімфатичні вузли, гістологічно уражені (частково змінена структура лімфатичного вузла) | |||
| Nx | Клінічно збільшені лімфатичні вузли без гістологічного ураження | |||
| М (внутрішні органи) | ||||
| М0 | Внутрішні органи не залучені | |||
| М1 | Внутрішні органи залучені | |||
| В (кров) | ||||
| В0 | Без циркулюючих атипових (Сезарі) клітин (або <5% лімфоцитів) | |||
| В 1 | Низьке пухлинне навантаження крові (≥5% лімфоцитів є клітинами Сезарі, але не В2) | |||
| В 2 | Високе пухлинне навантаження крові (≥100/мкл клітин Сезарі або підтверджена клональність) | |||
| Клінічна стадія | Класифікація TNMB | |||
| Т | N | M | B | |
| IA | T1 | N0 | M0 | B0–1 |
| IB | T2 | N0 | M0 | B0–1 |
| IIA | T1–2 | N1–2 | M0 | B0–1 |
| IIB | T3 | N0–2 | M0 | B0–1 |
| III | T4 | N0–2 | M0 | B0–1 |
| IVA1 | T1–4 | N0–2 | M0 | B2 |
| IVA2 | T1–4 | N3 | M0 | B0–2 |
| IVB | T1–4 | N0–3 | M1 | B0–2 |
Вибір тактики терапії залежить від стадії захворювання, типу ураження шкіри, віку, загального стану хворого, супутньої патології, а також попередньої історії лікування.
Для пацієнтів у ІА стадії з невеликими вогнищами ураження шкіри допускається тактика «спостерігай і чекай» під ретельним наглядом лікаря. При лікуванні пацієнтів з IA, IB та IIA стадіями можна використовувати топічні кортикостероїди (бетаметазон дипропіонат 0,05% або мометазону фуорат 0,1%), які наносяться на поверхню висипу 2 рази на добу до повної роздільної здатності [11]. Вузькохвильове ультрафіолетове опромінення (УФО) спектра В проводиться 2–3 рази на тиждень, рекомендоване за наявності плям і тонких бляшок. Гострі побічні ефекти — еритема, свербіж, відчуття печіння. При поширених та більш інфільтрованих елементах, фолікулярній формі грибоподібного мікозу застосовується ПУВА-терапія (псорален + ультрафіолетове опромінення (УФО) спектра А). За 2 год до опромінення пацієнт приймає перорально 8-метоксипсорален у дозі 0,6 мг/кг маси тіла, початкова доза опромінення залежить від типу шкіри (0,25–1,0 Дж/см2), потім з кожним сеансом доза підвищується на 0,25–0,5 Дж/см2 або більше залежно від еритеми. Лікування проводиться 3–4 рази на тиждень до зникнення висипу (30–35 сеансів). Загальна доза варіює в межах 50–80 Дж/см2, що є достатнім для досягнення клінічної ремісії. Побічні ефекти включають еритему, нудоту, свербіж, фотодерматит та фотокарциногенез [12–14]. Локальна променева терапія використовується у пацієнтів з невеликою кількістю висипу (10–20 Гр на курс). Разова вогнищева доза (РВД) — 1–1,2–1,5 Гр до сумарної вогнищевої дози (СВД) 30–40 Гр. Побічні променеві ушкодження, які виникають при цьому методі лікування, — еритема шкіри, часткова або повна алопеція, дистрофія нігтів, виражена сухість шкіри [15]. Також може використовуватися інтерферон-α (IFN-α) як препарат першої лінії для лікування хворих з IIB, III стадіями, ефективний у помірно високих дозах: 3–10 МО щодня або 3 рази на тиждень. IFN-α можна комбінувати з ПУВА, ретиноїдами та хіміотерапією [16].
За відсутності або недостатньої відповіді на лікування як другу лінію терапії при ранніх стадіях грибоподібного мікозу можна застосовувати ретиноїди (13-цис-ретиноєва кислота (ізотретиноїн, етретинат) у дозі 0,5–1 мг/кг маси тіла протягом 2–3 міс), які можуть призначатися в комбінації з ПУВА, IFN-α. Також застосовують інгібітори гістондеацетилаз (HDACi) (вориностат), проспідин: 100 мг/добу внутрішньом’язово або внутрішньовенно на курс 3–6 г, метотрексат призначають у дозах 25–75 мг на тиждень, можна комбінувати його з IFN-α.
Для лікування пізніх стадій (IIB-IVB) грибоподібного мікозу застосовують HDACi (вориностат), який призначається перорально по 400 мг щодня. Лікування проводять до досягнення відсутності ознак подальшого прогресування або до появи ознак неприйнятної токсичності [17, 18]. Електронно-променева терапія є вкрай ефективним методом лікування грибоподібного мікозу і може застосовуватися як на ранніх, так і пізніх стадіях як терапія першої лінії та при рецидивах/прогресуванні захворювання. Також застосовують режими системної хіміотерапії: CHOP, EPOCH, CMED/ABV, пентостатин, флударабін + IFN-α або циклофосфамід, гемцитабін [19, 20]. Алогенна трансплантація стовбурових клітин може бути рекомендована пацієнтам молодого віку на пізніх стадіях захворювання за відсутності ефекту при використанні інших видів терапії [21, 22].
Існують критерії визначення ефективності лікування при Т-клітинних лімфомах шкіри, які ґрунтуються на визначенні відповіді за шкірними покривами, лімфатичними вузлами.
Шкірні покриви
- Повна ремісія: 100% регресії висипу.
- Часткова ремісія: 50–99% регресії висипу від вихідного рівня, відсутність появи нових вузлів (Т3) у пацієнтів із Т1, Т2 або Т4 стадіями.
- Стабілізація захворювання: з <25% збільшення до <50% регресії висипу від вихідного рівня, відсутність появи нових вузлів (Т3) у пацієнтів із Т1, Т2 або Т4 стадіями.
- Прогресування захворювання: ≥25% збільшення висипу від вихідного рівня або поява нових вузлів (Т3) у пацієнтів із Т1, Т2 або Т4 стадіями, або відсутність відповіді — збільшення висипу від найнижчого рівня на 50% від вихідного рівня у хворих із досягнутою повною чи частковою ремісією.
- Рецидив: поява шкірного висипу у пацієнтів, які раніше досягали повної ремісії.
Лімфатичні вузли
- Повна ремісія: всі лімфатичні вузли ≤1,5 см у найбільшому діаметрі (довга вісь) або гістологічно негативні, також — лімфатичні вузли N3 та ≤1,5 см у найбільшому діаметрі та >1 см у найменшому діаметрі повинні бути ≤1 см у найменшому діаметрі або гістологічно негативними.
- Часткова ремісія: кумулятивне зниження ≥50% СДР (суми добутку максимального поздовжнього розміру і максимального поперечного розміру кожного ураженого лімфатичного вузла) та відсутність нових лімфатичних вузлів >1,5 см у діаметрі по довгій осі або >1 см по короткій осі.
- Стабілізація захворювання: відсутність критеріїв для повної і часткової ремісії та прогресування захворювання.
- Прогресування захворювання: підвищення ≥50% СДР від вихідних лімфатичних вузлів, або новий лімфатичний вузол >1,5 см у діаметрі по довгій осі, або >1 см по короткій осі, або відсутність відповіді: збільшення СДР на >50% від нижчого рівня у пацієнтів з частковою ремісією.
- Рецидив: поява нових гістологічно доведених N3 лімфатичних вузлів >1,5 см у найбільшому діаметрі.
Вісцеральні органи
- Повна ремісія: відсутність збільшення органа при фізикальному огляді та патологічних змін при томографії, біопсія будь-яких нових вогнищ, що з’явилися після лікування, для виключення лімфоми.
- Часткова ремісія: ≤50% регресії вогнищ печінки, селезінки або інших спочатку уражених органів за можливості виміряти обсяг ураження (СДР), відсутність збільшення органа в розмірах та залучення нових органів.
- Стабілізація захворювання: відсутність критеріїв для повної та часткової ремісії та прогресування захворювання.
- Прогресування захворювання: >50% збільшення органа у вигляді або ураження нового органа, або відсутності відповіді: збільшення СДР на >50% від нижчого рівня в пацієнтів з частковою ремісією.
- Рецидив: залучення у патологічний процес нового органа у пацієнтів із повною ремісією.
Кров
- Повна ремісія: В0 (див. табл. 2).
- Часткова ремісія: у хворих із стадією захворювання B2 — зниження кількісних параметрів ураження крові >50% від вихідного рівня.
- Стабілізація захворювання: відсутність критеріїв для повної та часткової ремісії та прогресування захворювання.
- Прогресування захворювання: B0 → B2 (див. табл. 2); або підвищення >50% від вихідного рівня (5000 пухлинних клітин/µL).
- Рецидив: підвищення рівня пухлинних лімфоцитів у крові у пацієнтів із повною ремісією ≥В1 (див. табл. 2).
Враховуючи критерії відповіді для шкіри, лімфатичних вузлів, вісцеральних органів та крові, визначається загальний критерій відповіді на лікування (табл. 3).
| Показник | Визначення | Шкіра | Лімфатичні вузлиКровВнутрішні органи |
| Повна ремісія (ПР) | Повне зникнення всіх клінічних симптомів хвороби | ПР | Усі категорії відповідають критеріям ПР/інтактні |
| Часткова ремісія (ЧР) | Регрес вимірюваних показників | ПР
ЧР |
Усі показники по органах та системах не відповідають ПР/інтактні та немає ознак ПБ
Для всіх категорій немає ознак ПБ, якщо будь-яка категорія залучена початково, принаймні одна з них має відповідати ПР чи ЧР |
| СБ (стабілізація захворювання) | Не досягнуто ПР, ЧР чи ПБ | ЧР
СБ |
У жодній категорії немає ознак ПБ, якщо будь-яка категорія залучена початково, у них не досягнуто ПР чи ЧР
ПР, ЧР, СБ у будь-якій категорії, в жодній немає ПБ |
| ПБ (прогресування захворювання) | Прогресування захворювання | ПБ | ПБ у всіх категоріях |
| Рецидив | Повторна поява хвороби за умови, що раніше було досягнуто ПР | Рецидив | Рецидив у всіх категоріях |
Синдром Сезарі становить менше ніж 5% від усіх первинних шкірних лімфом, середній вік пацієнтів становить 60–65 років, переважають пацієнти чоловічої статі.
Клінічно відзначаються генералізована еритродермія (рис. 1), лімфаденопатія, долонно-підошовний кератоз (рис. 2), оніходистрофії, алопеція.
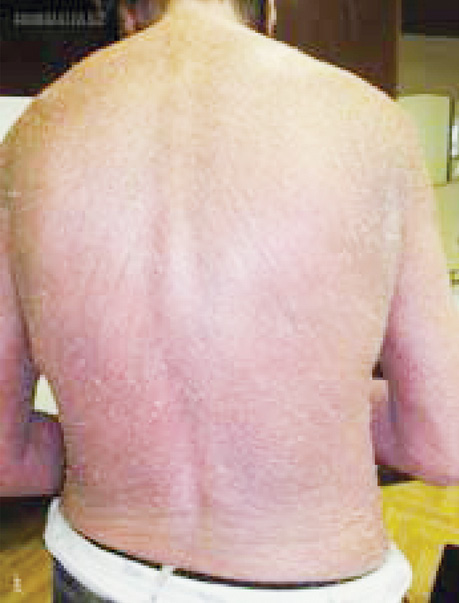
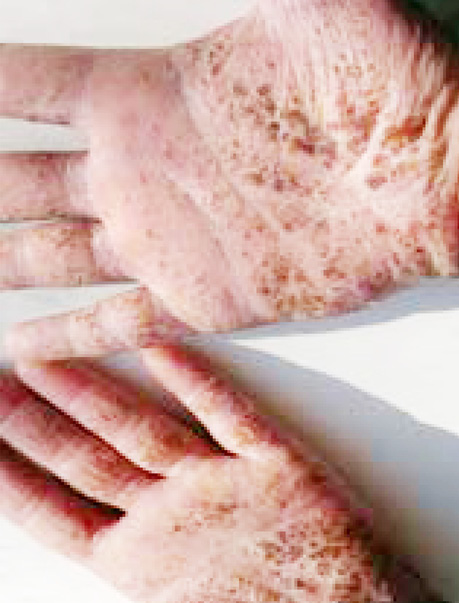
Методи обстеження хворих із синдромом Сезарі включають фізикальне обстеження (тип уражень шкіри, пальпація периферичних лімфовузлів, печінки, селезінки), біопсію підозрілих ділянок шкіри, біопсію лімфовузлів (за умови відсутності діагнозу після біопсії шкіри), імуногістохімічне дослідження CD CD4, CD5, CD7, CD8, CD20, CD30, CD25, CD56, TIA1, granzyme B, ßF1, TCR-CγM1, аналіз крові на наявність клітин Сезарі, молекулярно-генетичний аналіз біоптату шкіри, проточна цитометрія для клітин Сезарі (підрахунок CD4 +/CD7- та CD4+/CD26-), комп’ютерну томографію/позитронно-емісійну томографію голови, шиї, грудної, черевної порожнин та малого таза, дослідження кісткового мозку (аспірат/біопсія).
Для встановлення діагнозу синдрому Сезарі використовують такі критерії: відсутність попереднього грибоподібного мікозу, генералізована еритродермія (дифузна еритема, що покриває не менше 80% поверхні тіла з/без лущення), наявність у крові домінантного клону Т-лімфоцитів (визначається за допомогою полімеразної ланцюгової реакції (ПЛР) або Southern blot), наявність однієї або більше таких ознак — абсолютна кількість клітин Сезарі в крові ≥1000 клітин/мм3, підвищений вміст CD3+ або CD4+ клітин периферичної крові з коефіцієнтом співвідношення CD4/CD8 ≥10 (визначається за допомогою проточної цитометрії), підвищений вміст CD4+ клітин периферичної крові з аберантним імунофенотипом, що включає відсутність експресії 40% CD4+CD7– клітин) або CD26 (≥30% CD4+CD26– клітин).
Гістологічне, імуногістохімічне та молекулярно-біологічне (визначення реаранжування гена Т-клітинного рецептора методом ПЛР) дослідження шкіри та лімфатичних вузлів (у разі їх збільшення ≥1,5 см) є додатковими методами діагностики у незрозумілих діагностичних ситуаціях у пацієнтів із хронічною еритродермією невизначеної етіології.
Стадіювання синдрому Сезарі проводиться згідно з переглянутою TNM-класифікацією, запропонованою Міжнародним товариством з лімфом шкіри та Європейською організацією з вивчення та лікування раку (ISCL-EORTC staging system) (див. табл. 1).
При лікуванні синдрому Сезарі найбільш ефективним методом є екстракорпоральний фотоферез (ЕКФ), однак він не показаний пацієнтам з ураженням лімфовузлів та вісцеральних органів (стадії IVA та IVB). Сеанси проводять один раз на день протягом 2 днів з 4-тижневою перервою. ЕКФ добре переноситься хворими та не викликає виражених побічних явищ. IFN-α також може використовуватися як препарат першої лінії при синдромі Сезарі, він ефективний у більш високих дозах, ніж при грибоподібному мікозі: 9–18 МО щодня протягом 3 міс, потім 3 рази на тиждень. При непереносимості високих доз IFN-α препарат призначають у дозі 3–6 МО щодня або 3 рази на тиждень. Терапія низькими дозами метотрексату (<100 мг/тиждень) може призначатися як монотерапія за відсутності можливості інших видів лікування. Також можна комбінувати ці методи терапії.
Для лікування резистентних і рецидивних форм синдрому Сезарі застосовують хлорамбуцил у дозі 2–12 мг/добу у поєднанні з преднізолоном 20 мг/добу, пегільований ліпосомальний доксорубіцин у дозі 20–30 мг/м2 внутрішньовенно кожні 2–4 тиж, вориностат перорально по 400 мг щодня до відсутності ознак подальшого прогресування або ж до появи ознак неприйнятної токсичності, гемцитабін 1200 мг/м2 в 1-й, 8-й та 15-й день 28-денного циклу (3–6 курсів), поєднання флударабіну (25 мг/м2 кожні 3–4 тиж) та циклофосфаміду (250 мг/м2/добу 3 дні 1 раз на місяць) протягом 3–6 міс. Алогенна трансплантація кісткового мозку може розглядатися як потенційно можливий вид лікування у пацієнтів із синдромом Сезарі з агресивним перебігом та відсутністю ефекту від стандартних режимів терапії.
При синдромі Сезарі використовують такі самі критерії відповіді на лікування, як і при грибоподібному мікозі, які запропоновані ISCL, EORTC та Американським консорціумом з шкірних лімфом (USCLC).
Прогноз залежить від стадії патологічного процесу, середня 5-річна виживаність при цьому варіанті НХЛ становить 24%.
Первинні шкірні CD30+ лімфопроліферативні захворювання становлять 25% усіх первинних лімфом шкіри і є другою за частотою виникнення (після грибоподібного мікозу) патологією. Захворюваність у світі становить 0,1–0,2 випадку на 100 000 населення.
До первинних шкірних CD30+ лімфопроліферативних захворювань належать лімфоматоїдний папульоз та первинна анапластична CD30+ великоклітинна лімфома шкіри (АВКЛШ) [23].
Методи обстеження хворих з CD30+ лімфопроліферативними захворюваннями включають ретельний збір анамнезу (відзначають рецидивуючий папульозний висип, який мимовільно зникає, може бути згрупованим або дисемінованим (для лімфоматоїдного папульозу), попередні або супутні лімфопроліферативні захворювання (підвищення температури тіла >38 °С, профузна нічна пітливість, зменшення маси тіла більше ніж 10% за останні 6 міс)); фізикальний огляд (оцінка кількості та розмірів висипу, пальпація лімфовузлів), лабораторні дослідження (клінічний та біохімічний аналізи крові), радіологічне обстеження (при лімфоматоїдному папульозі рентгенографія грудної клітки, ультразвукове дослідження органів черевної порожнини та малого таза або комп’ютерна томографія для пацієнтів з відсутністю збільшених лімфатичних вузлів, гепатоспленомегалії та В-симптомів, при АВКЛШ: комп’ютерна томографія з контрастуванням); біопсія шкіри (гістологічне дослідження, імуногістохімічне дослідження, що включає такі маркери: CD3, CD4, CD5, CD7, CD8, CD20, CD30, ALK-1, EMA, CLA, CD56, TIA-1, granzim B, perforin), біопсія лімфовузлів (при збільшенні >1,5 см у діаметрі та/або щільній, нерівномірній консистенції трепанобіопсія кісткового мозку виконується при АВКЛШ у пацієнтів з множинним висипом та ураженням регіонарних лімфатичних вузлів).
Стадіювання первинних лімфом шкіри, відмінних від грибоподібного мікозу/синдрому Сезарі, проводиться згідно з рекомендаціями ISLE-EORTC.
Шкіра
- Т1 — поодинокий елемент шкірного висипу.
- Т1а — шкірний елемент <5 см у діаметрі, Т1b — шкірний елемент >5 см у діаметрі.
- Т2 — вогнищеве ураження шкіри: множинний висип, обмежений 1 зоною або 2 поряд розташованими зонами.
- Т2а — увесь висип розташовується в зоні <15 см у діаметрі.
- Т2b — увесь висип розташовується в зоні >15–<30 см у діаметрі.
- T2c — увесь висип розташовується в зоні >30 см у діаметрі.
- Т3 — генералізоване ураження шкіри.
- T3a — множинний висип, що займає зони, розташовані не поряд.
- T3b — множинний висип, що займає ≥3 зони.
Лімфатичні вузли
- N0 — немає збільшення периферичних та центральних лімфатичних вузлів, їх біопсія не потрібна.
- N1 — ураження 1 групи периферичних лімфатичних вузлів, що дренують ділянку теперішнього або попереднього шкірного висипу.
- N2 — ураження 2 або більше груп периферичних лімфатичних вузлів або ураження будь-яких периферичних лімфатичних вузлів, що не дренують ділянку теперішнього або попереднього шкірного висипу.
- N3 — ураження центральних лімфатичних вузлів.
Внутрішні органи
M0 — немає залучення внутрішніх органів.
M1 — залучення внутрішніх органів (з уточненням органа та морфологічним підтвердженням).
Клінічна картина при лімфоматоїдному папульозі характеризується наявністю рецидивуючого папульозного висипу, який мимоволі зникає, може бути згрупованим або дисемінованим. У деяких випадках папульозні елементи можуть розвиватися в некротичні виразки, що швидко ростуть, діаметром 1–4 см, які самостійно розрішуються з утворенням рубця (під мимовільним розрішенням розуміють спонтанну регресію кожного індивідуального елемента протягом тижнів або місяців, незалежно від появи нового висипу). Клінічна картина при АВКЛШ характеризується наявністю солітарного, згрупованого або множинного висипу, відсутністю клінічних ознак лімфоматоїдного папульозу, грибоподібного мікозу або інших Т-клітинних лімфом шкіри, а також відсутністю позашкірних вогнищ ураження.
Враховуючи хороший прогноз при лімфоматоїдному папульозі та високу частоту рецидивів практично після будь-якого виду терапії більшості пацієнтів пропонується тактика «спостерігай і чекай». В окремих випадках можна застосовувати кортикостероїди. При поширених та некротичних ураженнях застосовують ПУВА-терапію, низькі дози метотрексату. При прогресуванні застосовують IFN-α, anti-CD30 (Brentuximab) та Pralatrexate.
При лікуванні АВКЛШ з поодиноким чи локалізованими ураженнями застосовують радіотерапію, хірургічне лікування у поєднанні з променевою терапією або без. При мультифокальному ураженні використовують курси поліхіміотерапії за схемою CHOP, Iratumumab, Brentuximab, Bexarotene, IFN-α, Romodepsin, Vorinostat, Pralatrexate, препарати в рамках клінічних досліджень. За наявності мультифокального ураження та вісцеральних уражень застосовують курси поліхіміотерапії, які показані для лікування лімфом високого ступеня злоякісності, а також трансплантацію кісткового мозку [24].
ISCL, EORTC та Американським консорціумом з шкірних лімфом (USCLC) запропоновані наступні критерії відповіді на лікування при CD30+:
1. Шкіра
А. Лімфоматоїдний папульоз.
Повна ремісія: 100% зникнення висипу.
Часткова ремісія: 50–99% зникнення висипу від вихідного рівня, відсутність нового більшого вузликового висипу >2 см у діаметрі.
Стабілізація захворювання: від <50% збільшення до <50% зникнення висипу від вихідного рівня, відсутність нового більшого вузликового висипу >2 см у діаметрі.
Втрата відповіді: збільшення висипу від найнижчого рівня на 50% вихідного рівня у хворих із досягнутою повною чи частковою ремісією.
Прогресування захворювання: поява нового більшого і персистуючого вузликового висипу >2 см у діаметрі чи позашкірне поширення захворювання.
Рецидив: поява шкірного висипу у пацієнтів у стані повної ремісії захворювання.
В: АВКЛШ.
Повна ремісія: 100% зникнення висипу.
Часткова ремісія: 50–99% зникнення висипу від вихідного рівня, відсутність нових вузлів.
Стабілізація захворювання: від <25% збільшення до <50% зникнення висипу від вихідного рівня.
Прогресування захворювання: збільшення висипу більш ніж на 25% від вихідного рівня, або збільшення висипу від найнижчого рівня на 50% вихідного рівня у хворих із досягнутою повною чи частковою ремісією.
Рецидив: поява шкірного висипу у пацієнтів у стані повної ремісії захворювання.
2. Лімфатичні вузли
Повна ремісія: всі лімфатичні вузли <1,5 см у найбільшому діаметрі чи гістологічно негативні. Крім того, лімфатичні вузли, які на момент встановлення діагнозу були <1,5 см і при цьому гістологічно позитивні, повинні зменшитися до 1 см або бути негативними гістологічно.
Часткова ремісія: кумулятивне зниження ≥50% СДР та відсутність нових лімфатичних вузлів >1,5 см у діаметрі по довгій осі або >1 см по короткій осі.
Стабілізація захворювання: відсутність критеріїв для повної, часткової ремісії та прогресування захворювання.
Прогресування захворювання: підвищення ≥50% СДР від вихідних розмірів лімфатичних вузлів, або новий лімфатичний вузол >1,5 см у діаметрі по довгій осі або >1 см по короткій осі, або відсутність відповіді: збільшення СДР на >50% від найнижчого рівня у пацієнтів з частковою ремісією.
Рецидив: поява нових гістологічно доведених лімфатичних вузлів >1,5 см у найбільшому діаметрі у пацієнтів із повною ремісією.
3. Вісцеральні органи
Повна ремісія: відсутність збільшення органа при фізикальному огляді та відсутність патологічних змін при томографії, біопсія будь-яких нових вогнищ, що з’явилися після лікування, для виключення лімфоми.
Часткова ремісія: ≥50% регресії вогнищ печінки, селезінки або інших спочатку уражених органів при можливості виміряти обсяг ураження (СДР), відсутність збільшення органа в розмірах та залучення нових органів.
Стабілізація захворювання: відсутність критеріїв для повної, часткової ремісії та прогресування захворювання.
Прогресування захворювання: >50% збільшення органа у вигляді, чи ураження нового органа, чи відсутність відповіді: збільшення СДР на >50% від нижчого рівня у пацієнтів з частковою ремісією.
Рецидив: залучення нового органа у пацієнтів із повною ремісією.
На цей час залишається невирішеним питання, чи можуть при лімфатоїдному папульозі уражуватися лімфатичні вузли та вісцеральні органи. Виникнення CD30+ лімфопроліферативного процесу в лімфатичних вузлах та вісцеральних органах рекомендовано розцінювати як асоційовану з лімфоматоїдним папульозом вторинну анапластичну великоклітинну лімфому.
У більшості випадків лімфоматоїдний папульоз характеризується хронічним доброякісним перебігом і не впливає на виживання. АВКЛШ також має сприятливий прогноз з 5-річним виживанням 76–96%.
Підшкірна панікулітоподібна Т-клітинна лімфома становить 1% від усіх Т-клітинних лімфом шкіри та має індолентний перебіг: 19% пацієнтів — віком 20 років або молодше. Клінічно визначаються підшкірні вузлики, бляшки на ногах та руках, рідше — дифузні ураження. Спочатку захворювання має безсимптомний перебіг, але B-симптоми відмічають часто. 5-річне загальне виживання становить близько 82%.
До стандартних методів діагностики при підшкірній панікулітоподібній Т-клітинній лімфомі належать повне фізикальне обстеження, загальний аналіз крові, біохімічне дослідження крові, визначення рівня лактатдегідрогенази (ЛДГ), феритину в сироватці крові, β2-мікроглобуліну, ПЕТ/КТ шиї, грудної клітки, грудної клітки з контрастуванням, біопсія та аспірація кісткового мозку (потрібна завжди), біопсія шкіри з наступним гістологічним та імуногістохімічним дослідженнями.
Як терапію першої лінії застосовують преднізолон у монотерапії або в комбінації з циклофосфамідом. При рецидиві або рефрактерному перебігу використовують СНОР-режими, трансплантацію кісткового мозку, Romidepsin, нові препарати в рамках клінічних досліджень.
Екстранодальна NK/Т-клітинна лімфома, назальний тип відмічається рідко, характеризується швидким прогресуванням, агресивним перебігом. Клінічно визначаються пухлинні ураження у вигляді вузликів, які некротизуються і покриваються виразками; часта локалізація: обличчя, тулуб та кінцівки (рис. 3).
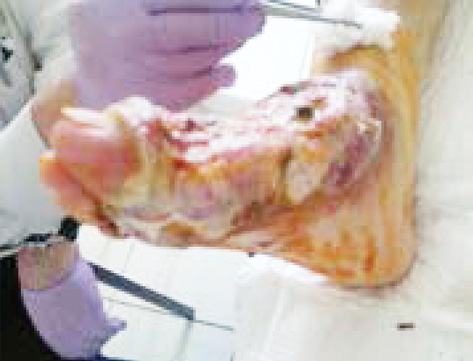
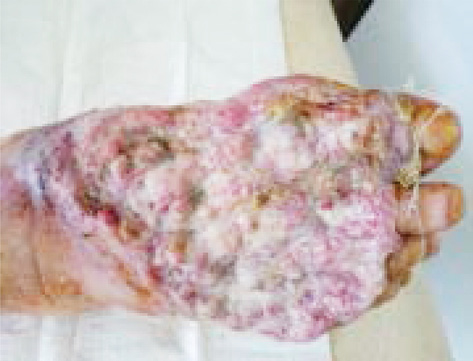
Для лікування екстранодальної NK/Т-клітинної лімфоми використовують курси хіміотерапії на основі L-аспарагінази (AspaMetDex, SMILE, GELOX) (рис. 4). Прогноз при цьому типі лімфоми несприятливий.
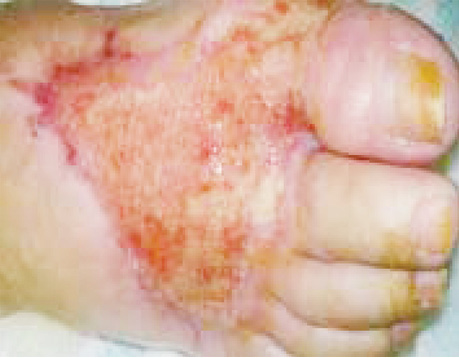
Первинна шкірна периферична Т-клітинна лімфома — неуточнена відмічається рідко. Клінічно визначаються поодинокі, локалізовані або генералізовані вузлики, плями. Лікування: поліхіміотерапія. Прогноз несприятливий, 5-річне виживання становить <20%.
Шкірна Y/δ Т-клітинна лімфома виникає рідко. Раніше належала до первинних шкірних периферичних Т-клітинних лімфом, NOS, у класифікації ВООЗ 2008 р. описується як окрема нозологія та рідкісний підтип CTCL. Пухлинні клітини експресують TCR-γδ, CD2, CD3, CD45RA, CD56 та цитотоксичні протеїни, CD4 та CD8 зазвичай «–». Прогноз поганий з медіаною виживання 18 міс. Для лікування шкірних Y/δ Т-клітинних лімфом застосовують ПХТ, а також алогенну трансплантацію.
CD8+ агресивна епідермотропна цитотоксична ШТКЛ відмічається рідко. У дебюті захворювання виявляють ерозивні або виразкові вузлики, бляшки, бородавчасті ураження, які можуть бути як локальними, так і дисемінованими. При цьому типі лімфоми відзначається швидка прогресія з ураженням лімфатичних вузлів, а також характерне системне залучення центральної нервової системи, яєчок, ротової порожнини, печінки, легень та часті коагулопатії. Середня виживаність хворих на CD8+ агресивну епідермотропну цитотоксичну ШТКЛ становить 32 міс. Стандарти терапії цього типу лімфом відсутні. При використанні ПХТ в більшості випадків можна досягти лише часткової відповіді. Застосовуються нові схеми терапії в рамках клінічних досліджень, алогенна трансплантація.
CD4+ дрібно-/середньоклітинна плеоморфна ШТКЛ виникає рідко. Клінічно відмічають появу одиничних вузликів, які частіше локалізуються в ділянці голови та шиї. Для CD4+ дрібно-/середньоклітинної плеоморфної ШТКЛ нехарактерні системні прояви. Прогноз сприятливий за наявності поодиноких чи кількох уражень, під час лікування яких застосовують хірургічне втручання, радіотерапію. При дисемінованих ураженнях шкіри відзначається несприятливий прогноз. Для лікування використовують протоколи терапії грибоподібного мікозу IIB–IV стадії.
Таким чином, діагностика первинних Т-клітинних шкірних лімфом потребує врахування багатьох факторів та використання широкого спектра сучасних методів обстеження (гістологічних, імунологічних, молекулярно-генетичних), які дають змогу правильно визначити прогноз та оптимізувати терапію. Лікування пацієнтів із первинними Т-клітинними лімфомами шкіри залежить від виду лімфоми та ступеня поширення процесу. Прогноз залежить від ступеня злоякісності пухлинного процесу, за деяких видів лімфом виживання становить до 20 років і більше, а за інших — до 1 року.
СПИСОК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
1. Willemze, R., Cerroni, L., Kempf, W., Berti, E., Facchetti, F., Swerdlow, S. H., & Jaffe, E. S. (2019). The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood, 133, 1703–1714. doi: 10.1182/blood-2018-11-881268.
2. Alaggio, R., Amador, C., Anagnostopoulos, I., Attygalle, A. D., Araujo, I. B. O., Berti, E., … Xiao, W. (2022). The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia, 36, 1720–1748. doi: 10.1038/s41375-022-01620-2.
3. Weinstock, M. A., & Gardstein, B. (1999). Twenty-year trends in the reported incidence of mycosis fungoides and associated mortality. American Journal of Public Health, 89, 1240–1244. doi: 10.2105/ajph.89.8.1240.
4. Chuang, T. Y., Su, W. P., & Muller, S. A. (1990). Incidence of cutaneous T cell lymphoma and other rare skin cancers in a defined population. Journal of the American Academy of Dermatology, 23, 254–256. doi: 10.1016/0190-9622(90)70208-y.
5. Kim, Y. H., Liu, H. L., Mraz-Gernhard, S., Varghese, A., & Hoppe, R. T. (2003). Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Archives of Dermatologу, 139, 857–866. doi: 10.1001/archderm.139.7.857.
6. Massone, C., Kodama, K., Kerl, H., & Cerroni, L. (2005). Histopathologic features of early (patch) lesions of mycosis fungoides: a morphologic study on 745 biopsy specimens from 427 patients. American Journal of Surgical Pathology, 29, 550–560. doi: 10.1097/01.pas.0000153121.57515.c6.
7. Smoller, B. R., Santucci, M., Wood, G. S., & Whittaker, S. J. (2003). Histopathology and genetics of cutaneous T-cell lymphoma. Hematology/Oncology Clinics of North America, 17, 1277–1311. doi: 10.1016/s0889-8588(03)00115-1.
8. Святенко, Т.В. (2007). Отёчные пятна и бляшки у женщины 68 лет. Дерматовенерология. Косметология. Сексопатология, 1–4(10), 389–392.
9. Kim, E. J., Hess, S., Richardson, S. K., Newton, S., Showe, L. C., Benoit, B. M., … Rook, A. H. (2005). Immunopathogenesis and therapy of cutaneous T cell lymphoma. Journal of Clinical Investigation, 115(4), 798–812. doi: 10.1172/JCI24826.
10. Kim, Y. H., Willemze, R., Pimpinelli, N., Whittaker, S., Olsen, E. A., Ranki, A., … Hoppe, R. T. (2007). TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood, 110, 479–484. doi: 10.1182/blood-2006-10-054601.
11. Zackheim, H. S. (2003). Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatologic Therapy, 16(4), 283–287. doi: 10.1111/j.1396-0296.2003.01639.x.
12. Diederen, P. V., van Weelden, H., Sanders, C. J., Toonstra, J., & van Vloten, W. A. (2003). Narrowband UVB and psoralen-UVA in the treatment of early-stage mycosis fungoides: a retrospective study. Journal of the American Academy of Dermatology, 48(2), 215–219. doi: 10.1067/mjd.2003.80.
13. Ponte, P., Serrão, V., & Apetato, M. (2010). Efficacy of narrowband UVB vs. PUVA in patients with early-stage mycosis fungoides. Journal of the European Academy of Dermatology and Venereology, 24(6), 716–721. doi: 10.1111/j.1468-3083.2009.03500.x.
14. Querfeld, C., Rosen, S. T., Kuzel, T. M., Kirby, K. A., Roenigk, H. H. Jr., Prinz, B. M., & Guitart, J. (2005). Long-term follow-up of patients with earlystage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Archives of Dermatologу, 141(3), 305–311. doi: 10.1001/archderm.141.3.305.
15. Hoppe, R. T. (2003). Mycosis fungoides: radiation therapy. Dermatologic Therapy, 16(4), 347–354. doi: 10.1111/j.1396-0296.2003.01647.x.
16. Olsen, E. A. (2003). Interferon in the treatment of cutaneous T-cell lymphoma. Dermatologic Therapy, 16(4), 311–321. doi: 10.1111/j.1396-0296.2003.01643.
17. Olsen, E. A., Kim, Y. H., Kuzel, T. M., Pacheco, T. R., Foss, F. M., Parker, S., … Duvic, M. (2007). Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology, 25(21), 3109–3115. doi: 10.1200/JCO.2006.10.2434.
18. Duvic, M., Olsen, E. A., Breneman, D., Pacheco, T. R., Parker, S., Vonderheid, E. C., … Geskin, L. J. (2009). Evaluation of the long-term tolerability and clinical benefit of vorinostat in patients with advanced cutaneous T-cell lymphoma. Clinical Lymphoma, Myeloma & Leukemia, 9(6), 412–416. doi: 10.3816/CLM.2009.n.082.
19. Scarisbrick, J. J., Child, F. J., Clift, A., Sabroe, R., Whittaker, S. J., Spittle, M., & Russell-Jones, R. (2001). A trial of fludarabine and cyclophosphamide combination chemotherapy in the treatment of advanced refractory primary cutaneous T-cell lymphoma. British Journal of Dermatology, 144(5), 1010–1015. doi: 10.1046/j.1365-2133.2001.04191.x.
20. Zinzani, P. L., Baliva, G., Magagnoli, M., Bendandi, M., Modugno, G., Gherlinzoni, F., … Tura, S. (2000). Gemcitabine treatment in pretreated cutaneous T-cell lymphoma: experience in 44 patients. Journal of Clinical Oncology, 18(13), 2603–2606. doi: 10.1200/JCO.2000.18.13.2603.
21. Olsen, E. A., Kim, Y. H., Kuzel, T. M., Pacheco, T. R., Foss, F. M., Parker, S., … Duvic, M. (2007). Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology, 25(21), 3109–3115. doi: 10.1200/JCO.2006.10.2434.
22. Duvic, M., Olsen, E. A., Breneman, D., Pacheco, T. R., Parker, S., Vonderheid, E. C., …, Geskin, L. J. (2009). Evaluation of the long-term tolerability and clinical benefit of vorinostat in patients with advanced cutaneous T-cell lymphoma. Clinical Lymphoma, Myeloma & Leukemia, 9(6), 412–416. doi: 10.3816/CLM.2009.n.082.
23. Ralfkiaer, E., Willemze, R., Paulli, M., & Kadin, M. E. (2008). Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In S. H. Swerdlow, E. Campo, N. L. Harris (Eds.), World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues (4th ed). Lyon: IARC Press.
24. Kempf, W., Pfaltz, K., Vermeer, M. H., Cozzio, A., Ortiz-Romero, P. L., Bagot, M., … Willemze, R. (2011). EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood, 118(15), 4024–4035. doi: 10.1182/blood-2011-05-351346.
Адреса для листування:
Алексик Олена Михайлівна
03022, Київ, вул. Здановської Юлії, 33/43
Державне некомерційне підприємство «Національний інститут раку»
E-mail: aleksikel@gmail.com
Сorrespondence:
Olena Aleksyk
33/43 Yulia Zdanovska str., Kyiv, 03022
Nonprofit Organization National Cancer Institute
E-mail: aleksikel@gmail.com





Leave a comment