




АL-амілоїдоз: труднощі та перспективи
Резюме. Summary. АL-амілоїдоз — це диспротеїноз, який уражає різні органи і системи, що призводить до загрозливої для життя органної недостатності. Захворювання відносно рідкісне, і суттєвою проблемою є його пізня діагностика, особливо амілоїдозу серця. AL-амілоїдоз розвивається в результаті позаклітинного відкладення фібрил, які складаються з моноклональних легких ланцюгів імуноглобулінів (співвідношення лямбда до каппа — 3:1). Навіть при невеликому клоні плазматичних клітин можливий розвиток масивного ураження органа даними відкладеннями. Цей стан може супроводжувати множинну мієлому або інші варіанти В-лімфопроліферативних захворювань і діагностуватися синхронно. Амілоїдоз часто не можуть діагностувати, оскільки ознаки і симптоми можуть імітувати симптоми більш поширених захворювань. За допомогою ранньої діагностики можливо запобігти прогресуючому пошкодженню органа. Коректне стадіювання і визначення прогностично значущих факторів до початку терапії надзвичайно важливі у контексті різних терапевтичних підходів і визначення стратегії в цілому для кожного пацієнта індивідуально. Недолік проспективних досліджень, особливо III фази, — одна з найбільших проблем в науці лікування AL-амілоїдозу. Режими терапії, які застосовуються для лікування пацієнтів з множинною мієломою, з різним ступенем успіху використовуються в якості терапії цього захворювання. У статті розглянуто сучасні підходи до діагностики та лікування пацієнтів з амілоїдозом легких ланцюгів імуноглобулінів, включаючи стандартні методи, а також ті, які наразі досліджуються. Виживаність при AL-амілоїдозі залежить від поширеності ураження, кількості органів, залучених до патологічного процесу. Найбільш значущим є наявність амілоїдозу серця, який розцінюється як несприятлива прогностична ознака. Однак з еволюцією підходів до лікування у значної частини пацієнтів вдається досягти успіху терапії. Оцінка гематологічної відповіді є ключовим фактором.
Резюме. АL-амилоидоз — это диспротеиноз, вовлекающий различные органы и системы, что приводит к жизнеугрожающей органной недостаточности. Заболевание относительно редкое, и существенной проблемой является поздняя диагностика, особенно амилоидоза сердца. AL-амилоидоз развивается в результате внеклеточного отложения фибрилл, которые состоят из моноклональных легких цепей иммуноглобулинов (соотношение лямбда к каппа — 3:1). Даже при небольшом клоне плазматических клеток возможно развитие массивного поражения органа данными отложениями. Это состояние может сопровождать множественную миелому или другие варианты В-лимфопролиферативных заболеваний и диагностироваться синхронно. Амилоидоз часто не диагностируется, поскольку признаки и симптомы могут имитировать симптомы более распространенных заболеваний. С помощью ранней диагностики возможно предотвратить прогрессирующее повреждение органа. Корректное стадирование и определение прогностически значимых факторов до начала терапии чрезвычайно важны ввиду различных терапевтических подходов и определения стратегии в целом для каждого пациента индивидуально. Недостаток проспективных исследований, особенно III фазы, — одна из самых больших проблем в науке лечения AL-амилоидоза. Режимы терапии, применяемые для лечения пациентов с множественной миеломой, з разной степенью успеха используются в качестве терапии данного заболевания. В статье рассмотрены современные подходы к диагностике и лечению пациентов с амилоидозом легких цепей иммуноглобулинов, включая стандартные методы, а также исследуемые в данное время. Выживаемость при AL-амилоидозе зависит от распространенности поражения, количества органов, вовлеченных в патологический процесс. Наиболее значимым является наличие амилоидоза сердца, который расценивается в качестве неблагоприятного прогностического признака. Однако с эволюцией подходов к лечению у значительной части пациентов удается достичь успеха терапии. Оценка гематологического ответа является ключевым фактором.
Получено 01.04.2019
Принято в печать 17.04.2019
DOI: 10.32471/clinicaloncology.2663-466X.38.22357
AL-амилоидоз — диспротеиноз, относящийся к гемобластозам. Показатель заболеваемости — 10 пациентов на 1 000 000 населения в год [1–5].
AL-амилоидоз — заболевание, при котором моноклональные легкие цепи иммуноглобулинов откладываются в тканях в виде парапротеинов. Идентифицировано более 30 различных специфичных белков, способных формировать фибриллы амилоида, из которых пять могут приводить к амилоидозу. С помощью диагностики на основе масс-спектрометрии постоянно выявляют все большее количество подвариантов заболевания. AL-тип амилоидоза встречается более чем у ¾ пациентов и вызван клоном плазматических клеток, количество которых в костном мозге у половины пациентов не превышает 10%. Клинические проявления разных вариантов амилоидоза неспецифичны, могут быть идентичными при разных вариантах амилоидоза и включают поражение сердца и почек, как сочетанное, так и изолированное, что возможно при любом из подтипов [1, 2, 5, 6]. Мишенями для отложения аномальных легких цепей иммуноглобулинов могут быть различные органы, однако выживаемость пациентов напрямую связана с выраженностью поражения сердца. При AL-амилоидозе наиболее часто поражается сердце, почки, реже — мягкие ткани, печень, периферическая и/или автономная нервная система, желудочно-кишечный тракт. Транстирениновый — ATTR-амилоидоз — это второй по частоте вариант амилоидоза, при котором также часто поражается сердечная мышца (табл. 1).
За последнее десятилетие улучшилось понимание патогенеза поражения органов амилоидом. С эволюцией знаний о данной патологии быстроменяющийся подход к лечению увеличил длительность жизни пациентов с этим заболеванием. Согласно различным данным общая 4-летняя выживаемость колеблется от 40 до 60% [1]. Однако все еще огромной проблемой остается ранняя смертность от распространенного сердечного поражения сердца. Основными причинами смерти пациентов с AL-амилоидозом являются прогрессирование сердечной недостаточности (СН) и внезапная сердечная смерть вследствие электромеханической диссоциации или жизнеугрожающих желудочковых аритмий [2, 3].
| Тип амилоида | Органы-мишени | |
| AL | Легкие цепи иммуноглобулинов | Сердце, почки, мягкие ткани, печень, периферическая и/или автономная нервная система, желудочно-кишечный тракт |
| ATTR | Транстирениновый (мутантный тип)Транстирениновый (дикий) | Периферическая и/или автономная нервная система, сердце, глаза, почки, лептоменингеальные оболочки |
| AA | Сывороточный амилоид А (SAA) | Сердце, связки, синовиальные оболочки |
| AApoAI | Аполипопротеин А-І (мутантный тип) | Почки, печень, сердце, щитовидная железа, автономная нервная система, желудочно-кишечный тракт |
| ALECT2 | Сывороточный белок, фактор хемотаксиса лейкоцитов | Почки, печень |
Клиническая картина и диагностика
Как упоминалось выше, при амилоидозе вовлекаться в патологический процесс могут несколько органов, и клиническая картина зависит от степени поражения амилоидом органа-мишени. Клиническая мимикрия связана с отсутствием специфических симптомов AL-амилоидоза.
К сожалению, первичный амилоидоз легких цепей иммуноглобулинов с поражением сердца диагностируется очень поздно. Почти в 40% случаев заболевание идентифицируют более чем через год после появления симптомов. Около 30% пациентов умирают в течение этого времени, несмотря на совершенствование подхода к терапии этого заболевания [6, 7].
Среди клинических проявлений амилоидоза легких цепей иммуноглобулинов доминируют: сильная усталость, потеря массы тела, ортостатическая гипотензия, одышка, отеки, протеинурия, карпальный туннельный синдром, периферическая нейропатия, автономная нейропатия, постуральная гипотензия, эректильная дисфункция, дисфункция мочевого пузыря/кишечника, гепатомегалия, макроглоссия, периорбитальная пурпура, «перемежающая хромота» нижней челюсти. Этот список может дополняться в зависимости от распространенности поражения амилоидом. Ранних признаков заболевания, к сожалению, нет. Но следует помнить о таргетной популяции пациентов с моноклональными гаммапатиями, которые подвержены развитию AL-амилоидоза. В качестве скрининговых обследований необходимо использовать биомаркеры сердечного и ренального поражения: N-концевой пронатрийуретический пептид тип-B (NT-proBNP), тропонин Т и/или I и суточную протеинурию. Эти обследования могут помочь снизить позднюю диагностику амилоидоза у пациентов с моноклональными гаммапатиями неопределенного генеза и нарушенным соотношением свободных легких цепей (free light chains — FLC) иммуноглобулинов.
Основные эхокардиографические признаки амилоидоза сердца: симметричная гипертрофия миокарда, преимущественно левого желудочка, дилатация предсердий, перикардиальный выпот, утолщение створок клапанов, зернистость миокарда [3]. Однако эти признаки обладают низкой точностью на ранних стадиях и не позволяют проводить дифференциальную диагностику с другими заболеваниями, которые сопровождаются концентрической гипертрофией левого желудочка. Для диагностики амилоидного поражения необходимо проводить комплексную эхокардиографическую оценку сердца, которая, помимо стандартной эхокардиографии в М- и 2D-режимах, включает анализ сегментарной систолической функции левого желудочка (продольная, циркулярная и радиальная деформация с помощью спекл-трекинг-эхокардиографии), анализ диастолической функции левого желудочка, а также оценку функции правого желудочка [3, 18]. В настоящее время установлена роль магнитно-резонансной томографии в диагностике амилоидоза сердца. Данное исследование входит в перечень обязательных согласно существующим рекомендациям (NCCN 2018). Метод магнитно-резонансной визуализации обладает высокой чувствительностью (88%), специфичностью (95%) для прижизненного выявления амилоидоза сердца, позволяет определить наличие зон с пораженными кардиомиоцитами вследствие накопления амилоида в межклеточном пространстве и дифференцировать их от неизмененного миокарда [8].
Установление диагноза требует морфологического подтверждения поражения амилоидом органа/ткани. Наиболее простым и доступным местом биопсии является жировая клетчатка передней брюшной стенки (метод обладает 80% чувствительностью). В случае отрицательного результата возможно проведение биопсии слизистой оболочки щеки, прямой кишки, нефробиопсии и т.д. Обязательным перед выполнением данной процедуры является оценка гемостаза. Особенно опасной в случае амилоидассоциированной коагулопатии может оказаться биопсия печени, которая может завершиться летальным кровотечением. Биопсия миокарда производится в случаях, когда есть подозрения на амилоидоз из неинвазивных тестов, но при проведении биопсии других тканей отложения амилоида не выявлены [9].
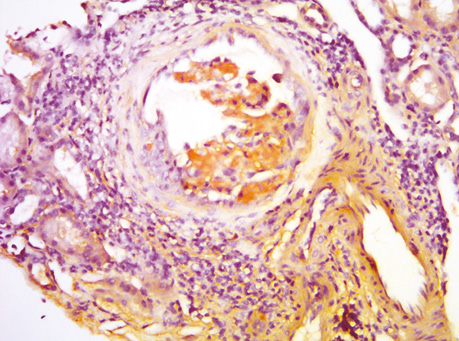
Характерным для амилоидоза является положительная окраска конго красным при морфологическом исследовании (рисунок). Однако следует помнить о том, что любые амилоидные отложения будут иметь положительную окраску с данным реактивом, а исключение других подтипов амилоидоза является критически важным. Метод флюоресцентной микроскопии и иммуногистохии с антителами к k (каппа) и λ (лямбда) цепям имеет достаточно высокую чувствительность и специфичность и в более чем 95% случаев позволяет правильно классифицировать амилоидоз. В случаях неинформативности световой микроскопии и иммуногистохимии может быть целесообразным метод масс-спектрометрии — дорогостоящей методики, при которой проводится лазерная микродиссекция конго красный-положительных областей слайдов, полученных из парафиноподобных блоков.
При достаточном материально-техническом оснащении проводят секвенирование генов для исключения или подтверждения возможного наследственного амилоидоза. Сцинтиграфия сердца с костными трассерами (Tc-3,3-дифосфоно-1,2-пропанодикарбоновой кислоты или пирофосфата) может дифференцировать AL-амилоидоз от транстиренинового амилоидоза (сильное поглощение), а также от ATTRwt (дикий тип транстиренинового амилоидоза, — ранее — сенильный системный амилоидоз), особенно у мужчин пожилого возраста [6].
Клон плазматических клеток в костном мозге при АL-амилоидозе, как упоминалось выше, часто небольшой, и обследование пациента должно включать иммунофиксацию сыворотки крови и мочи. FISH для определения наличия t (11;14) поможет выбрать терапевтический подход. Например, с наличием транслокации t (11;14) в костном мозге пациента повышается риск потенциальной неэффективности бортезомибсодержащих схем, а аномалия 1q21 говорит о рефрактерности к режиму M-dex (мелфалан + дексаметазон), преодолимой с помощью применения бортезомиба и аутологичной трансплантации стволовых клеток (АТСК).
Важное значение в диагностике амилоидоза имеет определение FLC иммуноглобулинов сыворотки крови, при котором распознают подтип AL-амилоидоза в зависимости от цепей, в сторону которых нарушено их соотношение (FLC-ratio) — λ или k. В 3 раза чаще диагностируют амилоидоз с гиперпродукцией λ-цепей. Разница между количеством вовлеченных и невовлеченных легких цепей (dFLC) имеет значение для стадирования и оценки гематологического ответа на терапию. Кроме того, этот показатель выступает в роли прогностического фактора. Определение dFLC в сочетании с NT-proBNP позволяет выделить пациентов с особенно неблагоприятным прогнозом [10].
В 2004 г. исследователями Клиники Мэйо (Mayo Clinic) была предложена система стадирования AL-амилоидоза, которая включала оценку уровня NT-proBNP и тропонина Т или I [7]. В 2012 г. она была усовершенствована и дополнена (табл. 2). Пороговые значения указанных маркеров повысились до 1800 нг/л для NT-proBNP и 0,025 нг/мл для тропонина Т, dFLC — 180 мг/л (Mayo Revised Criteria) [11]. При наличии клинико-морфологических, инструментальных (эхокардиография + стрейн, магнитно-резонансная томография) доказательств амилоидоза, нарушенном FLC-соотношении без превышения пороговых значений указанных маркеров диагностируется I стадия заболевания. В случае превышения у пациента порогового уровня по одному из маркеров устанавливается II стадия заболевания, двух — III и трех — IV соответственно. Для определения поздних стадий (III–IV) предложена Европейская система для распространенного кардиального амилоидоза, включающая 3 фактора: ≥III стадии согласно Mayo revised criteria, систолическое артериальное давление <100 мм рт. ст. и NT-proBNP >8500 нг/л. Подстадия a — это отсутствие данных факторов риска, в — наличие одного и с — двух. Данная система — очень удобная стратификационная шкала. Согласно результатам анализа выживаемости пациентов с разными стадиями доказано, что медиана выживаемости пациентов с I стадией превышала 90 мес, а для пациентов с III–IVC — около 3–6 мес.
| Система стадирования | Маркеры и пороговые значения | Стадия |
| Стандартная система Mayo | NT-proBNP >332 нг/л Тропонин Т >0,035 нг/мл (или тропонин І >0,01 нг/мл) |
I — ни один маркер не превышает пороговое значение II — один маркер превышает пороговое значение III — два маркера превышают пороговое значение |
| Европейская система для распространенного кардиального амилоидоза | >III стадия по стандартной системе Mayo Систолическое артериальное давление <100 мм рт. ст. NT-proBNP >8500 нг/л |
а — нет факторов риска в — один фактор риска с — два фактора риска |
| Пересмотренная система Mayo | NT-proBNP >1800 нг/л Тропонин Т >0,025 нг/мл dFLC >180 мг/л |
I — ни один маркер не превышает пороговое значение II — один маркер превышает пороговое значение III — два маркера превышают пороговое значение IV — три маркера превышают пороговое значение |
| Поражение почек | Скорость клубочковой фильтрации (СКФ) <50 мл/мин на 1,73 м Суточная протеинурия >5 г |
I — СКФ выше порогового значения, протеинурия — ниже порогового значения II — один из факторов риска присутствует III — СКФ ниже порогового, протеинурия — выше порогового значения |
Ключевыми показателями для интерпретации степени почечного поражения при амилоидозе являются: протеинурия >5 г/сут и СКФ <50 мл/мин. Принцип стадирования аналогичен описанному выше для оценки кардиального амилоидоза [11].
При вовлечении других органов и систем достаточно констатации его наличия. Стадирование проводится только по сердечно-сосудистой системе и почкам.
При подозрении на амилоидную нейропатию целесообразно проводить нейромиографию. При поражении дыхательной системы характерными будут рентгенологические изменения легочной ткани (множественные инфильтративные тени) и сниженные функциональные дыхательные тесты. Для поражения печени характерно повышение трансаминаз.
В контексте прогностических маркеров при амилоидозе изучена роль фактора дифференцировки роста 15 (GDF-15) — члена цитокинового суперсемейства трансформирующего фактора роста β (TGF-β). В совместной работе Центра исследования и лечения амилоидоза (Amyloid Research and Treatment Centre) в Павии, Италия, и клиники Афинского национального университета им. Каподистрии (National and Kapodistrian University of Athens), Греция, было доказано, что повышенный уровень GDF-15 в сыворотке крови (5658 или 7553 пг/мл) у пациентов с AL-амилоидозом почек ассоциируется с меньшей общей выживаемостью. GDF-15 оказался независимым прогностическим фактором и был предиктором диализа, не связанным со стадией почечного поражения согласно критериям Клиники Мэйо [12].
Лечение
На протяжении многих лет лечение AL-амилоидоза было исключительно химиотерапевтическим. Такая терапия направлена на уменьшение опухолевого клона плазматических клеток, сокращая количество цитотоксических легких цепей, наиболее тропных к кардиомиоцитам и клеткам мозгового вещества почек. Благодаря этому снижается органная дисфункция и повышается выживаемость. За последние годы были изучены таргетные терапевтические препараты, которые оказались мощным дополнением полихимиотерапии.
В основном режимы, применяемые для лечения первичного амилоидоза, были разработаны для терапии больных множественной миеломой. В основе выбора терапевтической тактики должна лежать риск-стратификация. Использование АТСК связано с неприемлемой токсичностью у пациентов с высоким риском. У пациентов группы низкого и промежуточного риска АТСК позволяет улучшить общую и безпрогрессивную выживаемость. К группе низкого риска при установлении диагноза относят пациентов, не имеющих таких факторов, как: NT-proBNP <5000 нд/л, кардиальный тропонин T <0,06 нд/мл, возраст <65 лет, ECOG 0–2, СКФ >50 мл/мин/1,73 м без применения диализа, СН <III функционального класса по NYHA, фракция выброса левого желудочка >45%, систолическое артериальное давление >90 мм рт. ст. (стоя), сниженная диффузионная способность легких (DLCO >50%).
Группу промежуточного риска составляют пациенты с I–IIIa стадией. Пациенты, у которых диагностированы амилоидоз сердца IIIb стадии и выше, а также СН ≥III функционального класса согласно NYHA, составляют группу самого неблагоприятного прогноза и требуют применения режимов с редукцией доз, а также бортезомибсодержащих схем (ввиду возможности достижения быстрого гематологического ответа) [19].
По данным Центра исследования и лечения амилоидоза в Павии, только 14% пациентов относятся к группе низкого риска, и им целесообразно планировать АТСК, 42% пациентов — промежуточного и 44% — высокого риска.
Высокодозовый мелфалан с последующей АТСК — одна из возможных опций в списке, рекомендуемом NCCN [4], однако отбор пациентов для интенсивного лечения должен быть очень тщательным во избежание смертности, связанной с лечением. У пациентов — кандидатов на АТСК данная опция позволяет существенно повысить общую выживаемость по сравнению со стандартными режимами полихимиотерапии. Наиболее высокие результаты эффективности лечения наблюдали у пациентов, достигших полного ответа, включая органный ответ. Эти данные были опубликованы исследователями Клиники Мэйо в 2007 г. на основании анализа лечения 282 пациентов. Важно, что только 36 пациентов получали циклофосфамид в качестве мобилизационного курса, а остальные 246 — только филграстим. 93 пациента достигли полного ответа, 108 — частичного и 81 не ответил на терапию. Трехлетняя общая выживаемость пациентов, достигших полного ответа, составила 90%, частичного — 74%, не ответивших на терапию — 30%. Снижение уровней FLC иммуноглобулинов после терапии коррелировало с уменьшением NT-proBNP сердечного биомаркера и улучшением выживаемости [13]. Из подвергшихся АТСК 72 пациента были под наблюдением длительное время и 43% из них прожили более 10 лет.
Режим Mel-Dex активно используется у категории пациентов, которым не показана АТСК. Согласно данным G. Palladini [20]мелфалан в комбинации с дексаметазоном в высоких дозах позволяет достичь гематологического ответа у 67% пациентов и у половины из них — полного ответа при медиане наблюдения 4,5 мес. У 48% этих больных удалось улучшить функцию органа, пораженного амилоидом. Полный ответ, достигнутый с помощью данного режима, возможно поддерживать у 70% пациентов на протяжении 3 лет. По данным автора, 50% этих больных достигли 5-летней медианы наблюдения.
В рамках работы French Myeloma Collaborative Group в рандомизированном исследовании сравнивали эффективность двух подходов к терапии первой линии: мелфалан + дексаметазон и высокодозовый мелфалан с последующей АТСК. И оказалось, что статистически значимой разницы в достижении гематологического и органного ответа не было [14].
В небольших когортных исследованиях проводилась оценка эффективности режимов комбинации альфа-интерферона с дексаметазоном и талидомида с дексаметазоном [15]. Во втором случае токсичность была существенно выражена. A.D. Wechalekar и соавторы показали эффективность режима CTD у 77 пациентов с распространенным системным амилоидозом, у 44 из которых развился рецидив заболевания. У большинства пациентов (74%) был достигнут гематологический ответ, у 21% — полный ответ. 8% пациентов не смогли продолжать лечение более 8 нед по причине выраженной токсичности. Уровень смертности, связанной с лечением, достигал 4% [16].
Имеется несколько сообщений относительно применения леналидомида в комбинации с дексаметазоном или циклофосфамидом и дексаметазоном в качестве второй линии терапии. Последняя комбинация достаточно эффективна у пациентов с III стадией и вовлечением трех и более органов и систем. Гематологический ответ наблюдался у 77% и органный — у 29%. Токсичность преимущественно гематологическая. Потенциальная нефротоксичность режима требует тщательного контроля и наблюдения пациента, хотя режим является амбулаторным [17].
Бортезомибсодержащие режимы — достаточно эффективная опция для пациентов с впервые диагностированным амилоидозом легких цепей иммуноглобулинов, а также с рецидивом заболевания.
Ответ на терапию оценивается согласно XI International Symposium on Amyloid and Amyloidosis [17] (табл. 3).
| Критерии ответа | Комментарии |
| Гематологический ответ | |
| Полный ответ | Негативный результат иммунофиксации сыворотки крови и мочи |
| Очень хороший частичный ответ | dFLC <40 мг/л |
| Частичный ответ | Снижение dFLC >50% исходного уровня |
| Нет ответа | Все остальные случаи |
| Кардиальный ответ | Снижение NT-proBNP на ≥30% и 300 нг/л (если исходно составлял >650 нг/л) или как минимум снижение на 2 балла степени СН согласно NYHA (если исходно III или IV ) |
| Почечный ответ | Уменьшение суточной протеинурии как минимум на 30% или снижение ее ниже 0,5 г/сут, при отсутствии почечной прогрессии — повышение СКФ >25% |
80% пациентов — потенциальных кандидатов на АТСК получают в первую линию режим CyBorDех. Биология клональной плазматической клетки при амилоидозе отличается от таковой при множественной миеломе, она все же экспрессирует поверхностный антиген CD38, что послужило основанием исследования с применением даратумумаба (анти-CD38 моноклонального антитела) у этой категории пациентов. V. Sanchorawala с коллегами опубликовали данные, подтверждающие эффективность и удовлетворительную переносимость даратумумаба у пациентов с рефрактерным амилоидозом после минимум одной линии терапии [6].
Выводы
Амилоидоз — агрессивное заболевание, поздняя диагностика которого ассоциируется с высоким уровнем ранней смертности. Своевременное выявление данного заболевания требует высокой степени настороженности разных специалистов (гематолога, кардиолога, нефролога, семейного врача).
Наличие СН и нефротического синдрома или «гипертрофии левого желудочка» при эхокардиографии в отсутствие согласующихся электрокардиографических данных должны вызывать подозрение на AL-амилоидоз. Существующие стратегии лечения, которые включают ингибиторы протеасом, значительно улучшили выживаемость в случае своевременного начала терапии. Моноклональные антитела против плазматических клеток (даратумумаб) и отложения легких цепей амилоида показали эффективность даже у предлеченных рефрактерных к терапии пациентов и требуют дальнейшего изучения.
Список использованной литературы
- Milani P., Merlini G., Palladini G. (2018) Novel therapies in light chain amyloidosis. Kidney Int. Rep., 3(3): 530–541.
- Palladini G., Merlini G. (2016) What is new in diagnosis and management of light chain amyloidosis? Blood, 128(2): 159–168.
- Несукай Е.Г., Даниленко А.А., Козлюк (2016) Амилоидоз: клиника, диагностика, лечение. Украинский кардиологический журнал, 5: 105–117.
- NCCN guidelines (2018) AL-amyloidosis (https://www.nccn.org/professionals/physician_gls/).
- Desport E., Bridoux F., Siraс C. (2012) AL amyloidosis. Orphanet J. Rare Dis., 7: 54.
- Sanchorawala V., Sarosiek S., Sloan J.M. (2017) Safety and tolerability of daratumumab in patients with relapsed light chain (AL) amyloidosis: preliminary results of a phase II study. Blood, 130: 507.
- Dispenzieri A., Gertz M.A., Kyle R.A. et al. (2004) Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J. Clin. Oncol., 22: 3751–3757.
- Karamitsos T.D., Piechnik S.K., Banypersad S.M. (2013) Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc. Imaging, 6(4): 488–497.
- Dispenzieri A., Kyle R., Merlini G. et al. (2009) International Myeloma Working Group guidelines for serum free light chain analysis in multiple myeloma and related disorders. Leukimia, 23: 215–224.
- Kumar S.K., Dispenzieri A., Lacy M.Q. et al. (2011) Changes in serum-free light chain rather than intact monoclonal immunoglobulin levels predicts outcome following therapy in primary amyloidosis. Am. J. Hematol., 86: 251–255.
- Palladini G., Milani P., Foli A. (2018) Presentation and outcome with second-line treatment in AL amyloidosis previously sensitive to nontransplant therapies. Blood, 131(5): 525–532.
- Kastritis E., Dimopoulos M. (2016) Recent advances in the management of AL amyloidosis. Bitish J. Hematol., 172(2): 170–186.
- Gertz M.A., Lacy M.Q., Dispenzieri A. et al. (2007) Effect of hematologic response on outcome of pathologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Hematologica, 92: 1415–1418.
- Palladini G., Merlini G. (2009) Current treatment of AL amyloidosis. Haematologica, 94(8): 1044–1048.
- Palladini G., Perfetti V., Perlini S. et al. (2005) The combination of thalidomide and intermediate dose dexamethasone in an effective but toxic treatment for patients with primary amyloidosis (AL). Blood, 105: 2949–2951.
- Wechalekar A.D., Goodman H.J., Lachmann H.J. (2007) Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood, 109(2): 457–464.
- Kumar S., Hayman S., Dispenzieri A. (2012) Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood, 119(21): 4860–4867.
- Karamitsos T.D., Piechnik S.K., Banypersad S.M. et al. (2013) Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. J. Am. Coll. Cardiol. Img., 6: 488–497.
- Kumar S., Dispenzieri A., Gertz M. (2012) Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol., 30(9): 989–995.
- Palladini G., Hegenbart U., Milani P. (2014) A staging system for renal outcome and early markers of renal response to chemotherapy in AL-amyloidosis. Blood, 124(15): 2325–2332.
Адрес:
Степанишина Яна Анатольевна
03022, Киев, ул. Ломоносова, 33/43
Национальный институт рака
E-mail: yankastep@gmail.com
Correspondence:
Stepanishyna Yana
33/43 Lomonosova Str., Kyiv 03022
National Cancer Institute, Kyiv
E-mail: yankastep@gmail.com





Leave a comment