




Клінічні рекомендації з діагностики та лікування нейроендокринних пухлин шлунка і дванадцятипалої кишки
Зубарєв М.Г., Колеснік О.О., Лукашенко А.В., Остапенко Ю.В., Бойко А.В.
Резюме. Нейроендокринні пухлини (НЕП) об’єднують доволі велику групу пухлин, які характеризуються біологічною різноманітністю та гетерогенністю. За даними Всесвітньої організації охорони здоров’я, за останні роки спостерігається підвищення частоти виявлення НЕП до 3,65 випадку на 100 тис. населення на рік. Стадія та диференціювання НЕП є основними факторами прогнозу у пацієнтів з НЕП шлунково-кишкового тракту (ШКТ). Застосування сучасних методів діагностики — комп’ютерної томографії, магнітно-резонансної томографії, сцинтиграфії, позитронно-емісійної томографії, поєднаної з комп’ютерною томографією, та імуногістохімічного дослідження біологічного матеріалу — відіграє ключову роль у виявленні НЕП ШКТ. Основні принципи хірургічного лікування полягають у адекватному обсязі лімфодисекції, видаленні всіх видимих первинних пальпабельних пухлин, усуненні рецидиву та метастазів. Медикаментозне лікування пацієнтів із НЕП ШКТ IV стадії залежить від ступеня градації (G), показників індекcу проліферації (Ki-67) та мітотичної активності і полягає у застосуванні пролонгованих октреотидів, імунотерапії, системної хіміотерапії та локорегіонарної терапії. Медіана загальної виживаності пацієнтів із НЕП ШКТ І–ІІ стадії, які отримали лікування, становить 112–170 міс, ІІІ — 70–105 міс та ІV — 13–60 міс відповідно. Представлені рекомендації з діагностики та лікування пацієнтів з НЕП ШКТ базуються на рекомендаціях ENETS 2016 р., NCCN 03/2017, дані яких оновлювалися на основі перегляду літератури на базі пошукового ресурсу PUBMED за 2015–2016 рр.

ВСТУП
Нейроендокринні пухлини (НЕП) об’єднують доволі велику групу пухлин, які характеризуються біологічною різноманітністю та гетерогенністю. Вперше термін карциноїдна пухлина (Karzinoide) впроваджений видатним німецьким вченим Оберндорфером у 1907 р. У свою чергу видатний гістолог М. Кульчицький відкрив так звані ентерохромафінні клітини (Kulchitsky cells), які виявилися ентеро-нейроендокринними за своєю природою. Ці клітини містяться серед інших епітеліальних клітин слизової оболонки шлунково-кишкового тракту (ШКТ) та є структурною ланкою дифузної ендокринної системи, продукуючи різноманітні гормони, які впливають на функціонування ШКТ. Власне з цим пов’язаний так званий карциноїдний синдром, який виникає у 20% випадків, його причиною є патологічна надмірна продукція різноманітних гормонів малігнізованими клітинами НЕП [1, 2].
Етіологія НЕП невідома. Доволі часто вони асоціюються з генетичними ендокринними синдромами Хіппеля — Ліндау, множинною ендокринною неоплазією (МЕН) та ін. Усі НЕП мають злоякісний потенціал.
Клінічною особливістю НЕП є латентний перебіг та їх повільне прогресування. Хоча, за даними Всесвітньої організації охорони здоров’я (ВООЗ), захворюваність на НЕП становить усього 2–3 випадки на 100 тис. населення на рік, останніми роками спостерігається підвищення частоти їх виявлення до 3,65 [3–5]. За даними літератури (NCCN), серед усіх НЕП приблизно третина локалізується у легенях та тимусі, а дві третини — у ШКТ.
Пацієнти з НЕП можуть мати симптоми, асоційовані з гормональною гіперсекрецією, а саме припливи, почервоніння шкіри обличчя, діарею при карциноїдному синдромі, гіпертензію при феохромоцитомі, а також прояви, спричинені гіперсекрецією інсуліну, глюкагону, гастрину та інших гормонів у випадку НЕП підшлункової залози. Пацієнтки з гормональними симптомами мають так звані функціонуючі НЕП або, відповідно, без симптомів — нефункціонуючі.
Безумовно, стадія та диференціювання НЕП є основними факторами прогнозу у пацієнтів із НЕП ШКТ. Існують ще декілька показників, які тією чи іншою мірою впливають на прогноз, а саме: стан краю резекції (R), васкулярна та периневральна інвазія [6, 7]. Функціональний статус НЕП також розцінюється як фактор прогнозу НЕП. Інші потенціальні чинники прогнозу: хромогранін А, підвищений його рівень або динаміка до підвищення в процесі лікування асоціюється з поганим прогнозом НЕП.
Підвищена експресія mTOR (мішень рапаміцину у комплексі з протеїнкіназою серин-треонінової специфічності — відповідає за клітинний ріст та виживання) асоціюється з коротшою медіаною загальної виживаності [8]. У НЕП тонкої кишки виявлено мутації інгібітора циклінзалежної кінази CDKN1B (p27), і власне втрата експресії CDKN1B є поганим прогностичним фактором [9, 10].
Циркулюючі пухлинні клітини також можуть бути прогностичним фактором, що базується на теорії про дисемінацію хвороби. У недавньому дослідженні виявлено, що, якщо питома вага циркулюючих клітин НЕП дорівнює або перевищує 1 циркулюючу пухлинну клітину в 7,5 мл крові, то це асоціюється з поганим прогнозом та гіршою медіаною безрецидивної та загальної виживаності [11].
Хірургічне лікування НЕП є ефективнішим порівняно медикаментозним методом. Так, наприклад, у випадку І та ІІ стадії нейроендокринних пухлин шлунка (Ш-НЕП) та тонкої кишки за умови радикального видалення НЕП з інтактним краєм резекції (R0) та дотримання інших онкологічних принципів 5-річна виживаність пацієнтів становить 73 та 100% відповідно. А при ІІІ та ІV стадії — 65 і 25% та 97 і 84% відповідно. У літературі є незначна кількість повідомлень щодо ефективності застосування ад’ювантної терапії у пацієнтів із ІІІ стадією G2–3 НЕП ШКТ, а саме схеми цисплатин або карбоплатин з етопозидом. Також описують і променеву терапію, яка може бути застосована у випадку нерезектабельних НЕП з G3 та високим індексом проліферації.
Основні принципи хірургічного лікування полягають в адекватному обсязі лімфодисекції, видаленні всіх видимих первинних пальпабельних пухлин (у випадку НЕП тонкої кишки), усуненні рецидиву, метастазів (Мтс), первинно нерезектабельних пухлини, які регресували на фоні терапії (можливо при ретельній селекції пацієнтів); у разі симптоматичних функціонуючих нерезектабельних або рецидивуючих НЕП та віддалених Мтс — виконання циторедуктивних операцій R2 (видалення >90%), планової холецистектомії у випадку лікування місцево-поширених НЕП з подальшою тривалою терапією пролонгованими октреотидами. Перед проведенням хірургічного лікування пацієнтам з функціонуючими НЕП слід парентерально призначити октреотид за добу до операції та після з метою профілактики карциноїдного кризу.
Медикаментозне лікування пацієнтів з НЕП ШКТ IV стадії залежить від ступеня градації (G), індексу проліферації (Ki-67) та мітотичної активності. Так, при високодиференційованих НЕП із низьким Ki-67 з успіхом застосовуються пролонговані октреотиди, такі як Сандостатин-ЛАР та ланреотид. Результати одержані у відомих дослідженнях PROMID та CLARINET, де у пацієнтів з Мтс НЕП ШКТ медіана безрецидивної виживаності становила 14,3 міс порівняно з 6 міс у групі плацебо [12].
Хіміотерапія не знайшла широкого застосування через невисоку ефективність. У пацієнтів із низькодиференційованими НЕП G3 та високим Ki-67 (>50%) проводиться хіміотерапія на основі платини, фторпіримідинів та ін. У сучасних дослідженнях виявлена відносна ефективність таргетної терапії деяких НЕП ШКТ (підшлункової залози) із застосуванням сунітинібу та еверолімусу (RADIANT-4), де медіана безрецидивної виживаності становила 11,5 міс проти 3,9 міс в групі плацебо.
Співпраця між спеціалістами різних напрямків — хірургів, діагностів, патоморфологів, радіологів — відіграє надзвичайно важливу роль у встановленні своєчасного правильного діагнозу НЕП та проведенні відповідного лікування.
Сучасні рекомендації з лікування пацієнтів з НЕП ШКТ базуються на рекомендаціях ENETS 2016 р., NCCN 03/2017, дані яких оновлювалися на основі перегляду літератури на базі пошукового ресурсу PUBMED за 2015–2016 рр.
Підготовлені клінічні рекомендації складаються з трьох частин, у яких наведено дані з епідеміології, прогнозу, діагностики та лікування НЕП ШКТ таких локалізацій: шлунка, дванадцятипалої кишки (ДПК) та тонкої кишки (у наступній нашій публікації).
І. Клінічні рекомендації з діагностики та лікування нейроендокринних пухлин шлунка
Ш-НЕП (в англомовній літературі — G-NETs), як і НЕП апендикса та тонкої кишки, виявляються найчастіше серед усіх інших НЕП ШКТ.
За епідеміологічними даними SEER (Surveillance, Epidemiology and End Results Program), представленими Національним інститутом раку США, частота виникнення Ш-НЕП становить 8,7% [3]. На відміну від інших НЕП, Ш-НЕП також характеризуються морфологічною різноманітністю та агресивністю. Клінічно ці пухлини найчастіше є безсимптомними та доброякісними, хоча спорадичні Ш-НЕП можуть імітувати карциному шлунка.
Слід відмітити, що за останні роки захворюваність на Ш-НЕП зросла [13–17]. Наприклад, вражаючі статистичні дані, представлені Великобританією, свідчать, що частота виникнення Ш-НЕП в період з 2000 по 2006 р. становила 0,16 та 0,15 на 100 тис. населення серед чоловіків та жінок відповідно. Таким чином, виявлено зростання захворюваності у 23 рази серед чоловіків та у 47 разів — серед жінок порівняно з даними за 1995 р. [13]. Подібне підвищення частоти захворюваності виявлено у США: з 0,03 (1973–1977 рр.) до 0,33 (2003–2007 рр.) на 100 тис. населення [2]. Японські дані теж свідчать про підвищення показників захворюваності на Ш-НЕП та НЕП дванадцятипалої кишки (ДПК-НЕП) серед населення з 1,05 (2005 р.) до 1,67 (2010 р.) на 100 тис. осіб [14, 18]. В європейських країнах також спостерігається зростання цього показника до 3,4 на 100 тис. населення [19]. За сучасними даними, стрімке підвищення захворюваності на Ш-НЕП та ДПК-НЕП пов’язують із доступністю та широким використанням ендоскопічних методів обстеження та поліпшенням інформованості й обізнаності лікарів і патоморфологів.
Класифікація Ш-НЕП. У 2010 р. ВООЗ представила класифікацію Європейської асоціації нейроендокринних пухлин (ENETS), засновану на мітотичній активності та ступені проліферації (індекс Ki-67) клітин НЕП — G (grade), як доповнення до вже наявної класифікації стадій НЕП (табл. 1) [3].
Таблиця 1. Класифікація Ш-НЕП за G
| ENETS градація | Мітотичний індекс |
Ki-67 індекс проліферації, % |
Класифікація ВООЗ 2010 |
| G1 | <2 | <2 | НЕП G1 (карциноїд) |
| G2 | 2–20 | 3–20 | НЕП G2 |
| G3 | >20 | >20 | НЕП карцинома G3, великоклітинний або дрібноклітинний тип |
Високодиференційовані Ш-НЕП поділяються на три типи (табл. 2) [20–31].
Таблиця 2. Характеристики Ш-НЕП
| Характеристики | І тип | ІІ тип | ІІІ тип |
| Частота серед всіх Ш-НЕП | 70–80% | 5–10% | 10–15% |
| Асоційовані хвороби | Хронічний атрофічний гастрит |
МЕН І типу/синдром Золлінгера — Еллісона (СЗЕ) |
− |
| Розподіл за статтю | Більше жінок | Жінки=чоловіки | Більше чоловіків |
| Кількість пухлин | Множинні | Множинні | Солітарні |
| Розмір | <10 мм | <10 мм | >10 мм |
| Локалізація | Дно та тіло | Дно та тіло | Будь-яка |
| Гістологічна градація | G1 | G1/2 | G3 |
| Ступінь інвазії | Слизова/підслизова | Слизова/підслизова | Будь-яка |
| Рівень гастрину | Високий | Високий | Норма |
| рН шлунка | Високий | Низький | Норма |
| Ризик метастазування | 2–5% | 10–20% | >50% |
| Прогноз | Добрий | Посередній | Поганий |
Ш-НЕП І типу об’єднують 70–80% всіх Ш-НЕП та виникають найчастіше у жінок, є асимптоматичними та переважно випадково виявляються при ендоскопічному обстеженні. Гістологічно цей тип складається з ентерохромафіноподібних клітин (enterochromaffinlike — ECL), інакше Ш-НЕП І типу називають ECLомами (ECLomas) [29]. Найчастіше діагностують у пацієнтів із хронічним атрофічним гастритом, у тому числі аутоімунним та хелікобактерасоційованим гастритом, відповідно, у них також виявляють хронічну гіпергастринемію. У Ш-НЕП І типу імуногістохімічний аналіз визначає позитивний хромогранін А, синаптофізин, VMT (vesicular monoamin transporter) та соматостатиновий рецептор 2А.
Найчастіше Ш-НЕП І типу дрібні й множинні, локалізуються у тілі та дні шлунка, обмежені слизовою оболонкою або підслизовим шаром стінки шлунка. Оскільки ці пухлини у більшості випадків G1, їхній потенціал до метастазування дуже низький і тому прогноз вважається дуже добрим. Слід зауважити, що пацієнти з І типом Ш-НЕП, асоційованим з аутоімунним гастритом, можуть мати інші аутоімунні захворювання, такі як цукровий діабет І типу, аутоімунний тиреоїдит та первинний біліарний цироз [30].
Ш-НЕП ІІ типу становлять 5–6% серед усіх Ш-НЕП і мають подібні характеристики за розміром та розповсюдженістю з І типом. Однак виникають в однаковій пропорції у жінок і чоловіків та нерідко мають клінічну картину пептичної виразки. ІІ тип асоціюється з множинною ендокринною неоплазією І типу та СЗЕ [32, 33]. Майже 30% Ш-НЕП ІІ типу метастазують, що спричиняє зниження виживаності пацієнтів та гірший прогноз порівняно з хворими з І типом.
ІІІ тип становить близько 10–15% усіх Ш-НЕП, такі пухлини найчастіше спорадичні, виникають незалежно від рівня секреції гастрину та не асоціюються з ECL гіперплазією. Клінічно проявляються як одиничні пухлини розміром більше 10 мм, G3, інфільтрують м’язовий шар, метастазують у печінку та регіонарні лімфатичні вузли (ЛВ), з анемією, втратою апетиту, диспепсією, ШКТ-кровотечею та зменшенням маси тіла [25].
Карциноїдний синдром виникає у близько 1% пацієнтів із Ш-НЕП, найчастіше ІІІ типу з наявними Мтс у печінці, і проявляється такими симптомами: раптове почервоніння шкіри та припливи, тахікардія, діарея [34].
Стадіювання Ш-НЕП за системою TNM
Первинна пухлина (Т):
- Tх — первинна пухлина не може бути оцінена;
- T0 — немає даних про первинну пухлину;
- Тіs — карцинома in situ/дисплазія (розмір пухлини <0,5 мм), не пов’язана зі слизовою оболонкою;
- T1 — пухлина поширюється за lamina propria або у підслизову оболонку, розміром <1 см;
- T2 — пухлина поширюється у м’язову оболонку або розміром >1 см;
- T3 — пухлина поширюється субсерозно;
- T4 — пухлина поширюється на вісцеральну очеревину/серозну оболонку або інші органи/сусідні структури.
За наявності декількох пухлин до T слід додати (m).
Регіонарні ЛВ (N):
- Nх — регіонарні ЛВ неможливо оцінити;
- N0 — Мтс у регіонарних ЛВ не виявляються;
- N1 — Мтс у регіонарних ЛВ.
Віддалені Мтс (М):
- M0 — відсутні віддалені Мтс;
- М1 — віддалені Мтс (табл. 3).
Таблиця 3. Стадіювання Ш-НЕП за системою TNM (7-е видання 2010 р.)
| Стадія | T | N | M |
| 0 | is | 0 | 0 |
| І | 1 | 0 | 0 |
| ІІА | 2 | 0 | 0 |
| ІІВ | 3 | 0 | 0 |
| ІІІА | 4 | 0 | 0 |
| ІІІВ | 1–4 | 1 | 0 |
| IV | 1–4 | 0–1 | 1 |
Прогноз та показники виживаності. За світовими даними та SEER, при Ш-НЕП І типу прогноз доволі позитивний. Так, при проведенні ендоскопічного спостереження та резекції великих за розміром НЕП (>10 мм) безрецидивна виживаність пацієнтів становить близько 24 міс та може бути досягнута 100% загальна виживаність.
За даними SEER (2008 р.) визначено наступний розподіл показників виживаності хворих на Ш-НЕП G1–2 залежно від розповсюдженості первинної пухлини, наявності регіонарних та віддалених Мтс:
- первинна пухлина локальна (T1–3N0M0) — медіана загальної виживаності 163 міс, 3-річна — 80%, 5-річна — 73%, 10-річна — 56%;
- наявність Мтс у регіонарних ЛВ та місцеве розповсюдження пухлини (T1–4N1M0) — медіана загальної виживаності 76 міс, 3-річна — 75%, 5-річна — 65%, 10-річна — 43%;
- наявність віддалених Мтс (T1–4N1M1) — медіана загальної виживаності 13 міс, 3-річна — 33%, 5-річна — 25%, 10-річна — 9% [35].
Діагностика Ш-НЕП. Ендоскопічна гастродуоденоскопія з прицільною оцінкою пухлини та слизової оболонки шлунка залишаються золотим стандартом діагностики Ш-НЕП.
На рис. 1 надано зображення, виконані при проведенні ендоскопічної гастродуоденоскопії із застосуванням спеціальних режимів зображення та ендоскопічного ультразвукового дослідження (УЗД). На доповнення до рис. 1 слід додати, що іноді НЕП на поверхні можуть мати втягнення у центрі, яке свідчить про підслизовий ріст пухлини. Ш-НЕП ІІІ типу найчастіше солітарні, великі за розміром (>20 мм), з виразкуванням на поверхні, яке свідчить про глибоку інвазію [29, 36].
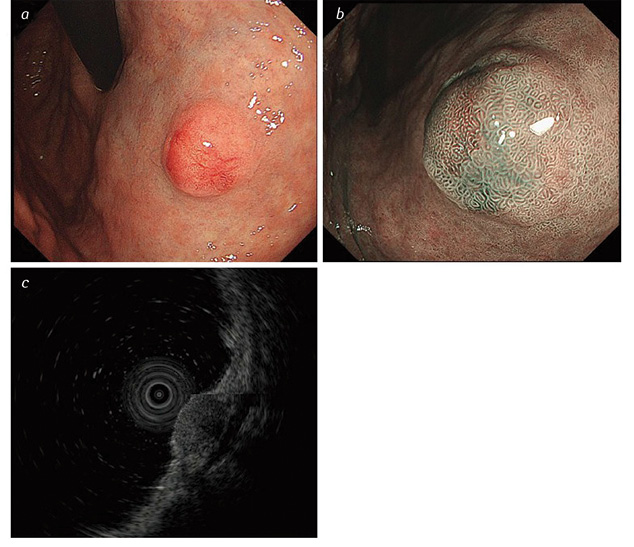
Ендоскопічне ультразвукове дослідження є важливим методом діагностики Ш-НЕП, надає інформацію про глибину пухлини та її локалізацію у стінці шлунка. За даними ендоскопічного УЗД, Ш-НЕП найчастіше розташовані у другому (глибоко у слизовій оболонці) або у третьому (підслизово) ехошарі та мають вигляд гіпоехогенної інтрамуральної структури [37].
Комп’ютерна томографія (КТ) та магнітно-резонансна томографія (МРТ) успішно використовуються для виявлення Мтс НЕП та її локального розповсюдження. Роль традиційної позитронно-емісійної томографії, поєднаної з комп’ютерною томографією (ПЕТ-КТ), у діагностиці Ш-НЕП залишається не визначена. Хоча за сучасними світовими даними встановлено, що ПЕТ-КТ із застосуванням трейсерів, мічених Ga-68 (Ga-DOTATOC, -DOTANOC та DOTATATE), має високу специфічність і чутливість при виявленні НЕП, але потребує подальших досліджень для використання у випадку Ш-НЕП [38–40].
Соматостатин-рецепторна сцинтиграфія — СРС (октреоскан) — рекомендована з метою виявлення Мтс НЕП у ЛВ, печінці, кістках [3].
Визначення рівня гастрину у крові пацієнтів із Ш-НЕП має значення при Ш-НЕП І та ІІ типів, оскільки у цих випадках його рівень підвищений. Однак цей показник не є специфічним.
Лікування Ш-НЕП. Стратегія лікування пацієнтів із Ш-НЕП залежить від типу, розміру, наявності факторів ризику, таких як інфільтрація м’язового шару, збільшення клітинної проліферації та метастазування.
Ш-НЕП І типу. Рекомендовано застосування консервативних методів ведення пацієнтів, а саме ендоскопічного спостереження у разі, якщо розмір пухлини <10 мм і немає ознак інвазії підслизового шару [31, 45, 46].
Ендоскопічна резекція (ЕР) може бути застосована за наявності пухлини розміром <20 мм (перевага надається солітарній пухлині), без ознак інвазії у м’язову оболонку, без Мтс. Останні дослідження підтверджують, що ендоскопічне субмукозне видалення Ш-НЕП є більш ефективним порівняно з традиційним мукозним видаленням (як при поліпектомії), більш радикальним із нижчою частотою рецидивів [43, 44, 60–70].
Резекція стінки шлунка (лапароскопічно/відкрито) може бути рекомендована у таких випадках, як: пухлина розміром >20 мм, множинні пухлини, поширення за межі підслизової оболонки, наявність Мтс у ЛВ або інших органах, низькодиференційовані пухлини [3].
Антрумектомія розглядається як варіант лікування при рецидиві Ш-НЕП після ЕР та гіпергастринемії і може бути виконана лапароскопічно [47, 48]. Але цей метод не може знизити ризик метастазування та подальших рецидивів захворювання [3, 47].
При рецидивах після ендоскопічного лікування та локальних резекцій необхідно проводити більш розширене хірургічне лікування, а саме резекцію шлунка (парціальну/субтотальну) з лімфаденектомією.
Медикаментозне лікування при Ш-НЕП І типу є обмеженим. Інгібітори соматостатинових рецепторів (ІСР) (ланреотид, пролонгований октреотид), які пригнічують секрецію гастрину G-клітинами та знижують гіперплазію ECL клітин, є також ефективними препаратами для зменшення кількості та розміру первинних Ш-НЕП [50–53]. Але їх рутинне застосування не рекомендоване, особливо у ранній стадії захворювання. ІСР можна успішно застосовувати у випадку рецидивних Ш-НЕП І типу, при множинних пухлинах та у тих пацієнтів, у яких не застосовували ЕР [54–56]. Також ІСР застосовують у хворих з Мтс, які мають експресію соматостатинових рецепторів та низький Ki-67 (<5%).
Антагоніст гастринових рецепторів — нетазепід — у декількох сучасних дослідженнях, маючи антипроліферативні властивості, показав свою ефективність, зменшуючи розмір та кількість Ш-НЕП І типу [57, 58]. Але рутинно не застосовується.
Ш-НЕП ІІ типу. Лікування хворих на Ш-НЕП ІІ типу практично не відрізняється від такого при І типі [36]. Відмінність полягає лише у більш агресивному хірургічному підході. Локальна резекція стінки шлунка, ураженої НЕП, залишається методом вибору. Цей підхід пов’язаний з більш високим метастатичним потенціалом Ш-НЕП ІІ типу порівняно з І типом [30]. Висока частота виявлення інших НЕП у ДПК та підшлунковій залозі у хворих на Ш-НЕП ІІ типу зумовлює застосування хірургічного підходу [53].
Ш-НЕП ІІІ типу. Лікування хворих на Ш-НЕП ІІІ типу проводиться хірургічно із застосуванням таких самих принципів, як при карциномі шлунка (резекція шлунка/гастректомія, лімфаденектомія D1) [3, 42, 70].
Спостереження. Пацієнтам з НЕП розміром <20 мм, з Ш-НЕП І та ІІ типу після лікування кожні 6–12 міс протягом 3 років проводять езофагогастродуоденоскопію (ЕГДС). Після 3 років — ЕГДС щороку. Згодом — кожні 2 роки.
Хворим на Ш-НЕП ІІІ типу або НЕП >20 мм І та ІІ типу після лікування проводити ЕГДС кожні 3 міс до 1 року та кожні 6 міс наступного року. Згодом — 3 роки щорічно, потім кожні 2 роки.
Такі обстеження, як УЗД, КТ та МРТ, можуть бути застосовані за потреби у кожному конкретному випадку [37, 42].
ІІ. Клінічні рекомендації з діагностики та лікування нейроендокринних пухлин дванадцятипалої кишки
ДПК-НЕП (у англомовній літературі — D-NETs) — доволі рідкісні. Захворюваність на ДПК-НЕП дещо зросла за останні роки і становить, за даними SEER, 0,19 на 100 тис. населення. 3% випадків ДПК-НЕП є первинними, серед НЕП тонкої кишки первинні утворення налічують 11% та 8% — серед усіх НЕП ШКТ. Вони частіше виникають у чоловіків, ніж у жінок, на відміну від Ш-НЕП [72–75].
Клінічні прояви ДПК-НЕП. Більшість ДПК-НЕП локалізуються у цибулині та низхідній частині ДПК, 20% із них розташовані у періампулярній зоні [76, 77]. Не зважаючи на широкий спектр різноманітних гормонів, які продукують ДПК-НЕП, 90% з них нефункціональні [73]. Тому ДПК-НЕП в основному випадково виявляють при ЕГДС. Симптоми, які належать до СЗЕ, наявні у 10% всіх ДПК-НЕП. Карциноїдний синдром виявляють у 3% пацієнтів з ДПК-НЕП [35]. ДПК-НЕП найчастіше солітарні, малі за розміром (75% <20 мм). Наявність множинних ДПК-НЕП, які виявляються у 9–13% випадків, мають насторожувати клініцистів щодо можливої наявності у пацієнта МЕН І типу та СЗЕ, оскільки доведено, що 60–95% із них можуть мати асоційовану гастриному [78, 79].
Також слід звернути увагу на те, що ДПК-НЕП у 60–90% хворих метастазують у ЛВ та у 10% випадків — у печінку, хоча первинні пухлини обмежені слизовою оболонкою або підслизовим шаром [80–84].
Прогноз та показники виживаності. П’ятирічна виживаність пацієнтів з високодиференційованими ДПК-НЕП становить 80–85% [74, 84].
За даними SEER (2008 р.) визначено наступний розподіл показників виживаності хворих на ДПК-НЕП залежно від розповсюдженості первинної пухлини, наявності регіонарних та віддалених Мтс:
- первинна пухлина локальна (T1–3N0M0) — медіана загальної виживаності 112 міс, 3-річна — 80%, 5-річна — 68%, 10-річна — 48%;
- наявність Мтс у регіонарних ЛВ та місцеве розповсюдження пухлини (T1–4N1M0) — медіана загальної виживаності 69 міс, 3-річна — 75%, 5-річна — 55%, 10-річна — 44%;
- наявність віддалених Мтс (T1–4N1M1) — медіана загальної виживаності 57 міс, 3-річна — 60%, 5-річна — 46%, 10-річна — 27% [71].
Стадіювання ДПК-НЕП за TNM, 7-ме видання, 2010 р.
Первинна пухлина (Т):
- Tх — первинна пухлина не може бути оцінена;
- T0 — немає даних про первинну пухлину;
- T1 — пухлина поширюється за lamina propria або у підслизову оболонку, розміром <1 см (стінка кишки), пухлина <1 см — ампула;
- T2 — пухлина поширюється у м’язову оболонку або розміром >1 см (стінка кишки)/пухлина >1 см в ампулі;
- T3 — пухлина поширюється субсерозно без залучення інших органів або структур;
- T4 — пухлина поширюється на вісцеральну очеревину/серозну оболонку або інші органи/сусідні структури.
За наявності декількох пухлин до T слід додати (m).
Регіонарні ЛВ (N):
Nх — регіонарні ЛВ неможливо оцінити;
N0 — Мтс у регіонарних ЛВ не визначаються;
N1 — Мтс у регіонарному ЛВ.
Віддалені Мтс (М):
- M0 — відсутні віддалені Мтс;
- М1 — віддалені Мтс (табл. 4).
Таблиця 4. Стадіювання ДПК-НЕП за системою TNM (7-ме видання 2010 р.)
| Стадія | T | N | M |
| 0 | is | 0 | 0 |
| І | 1 | 0 | 0 |
| ІІА | 2 | 0 | 0 |
| ІІВ | 3 | 0 | 0 |
| ІІІА | 4 | 0 | 0 |
| ІІІВ | 1–4 | 1 | 0 |
| IV | 1–4 | 0–1 | 1 |
Гістологічна класифікація ДПК-НЕП [3, 16, 85]
Класифікація ВООЗ, яка базується на G:
- G1 — зустрічається у 50–75% пацієнтів;
- G2 — 25–50%;
- G3 (нейроендокринна карцинома) — <3%.
ДПК-НЕП поділяються на 5 підтипів:
- дуоденальна гастринома — 50–60% випадків;
- соматостатин-продукуючі НЕП — 15%;
- нефункціонуючі серотонінвмісні НЕП — 19–27%;
- низькодиференційовані нейроендокринні карциноми — <3%;
- гангліоцитарні парагангліоми <2%.
Дуоденальні гастриноми у більшості випадків спорадичні або пов’язані з МЕН І типу і СЗЕ [23]. Останні дані свідчать, що гастриноми етіологічно можуть асоціюватися з H. pylori з атрофічним гастритом або при тривалому використанні інгібіторів протонної помпи [86, 87]. ДПК-НЕП майже завжди невеликих розмірів, 70–85% з них розміщені переважно у першій або другій частині ДПК, у так званому гастриномічному трикутнику, який локалізований у правому верхньому квадранті й обмежений ДПК та головкою підшлункової залози [73].
Так звані сімейні ДПК-НЕП, асоційовані з МЕН І типу, найчастіше бувають множинними, на відміну від інших ДПК-НЕП. Ці НЕП нерідко важко діагностувати [85]. Незважаючи на їхні невеликі розміри, доволі часто виявляють Мтс у ЛВ, а у 5–10% хворих присутні Мтс у печінці [80, 89].
Соматостатин-продукуючі пухлини (соматостатиноми) переважно розташовані в ампулі або періампулярній зоні, 20–30% з них пов’язані з нейрофіброматозом 1-го типу [90]. Ці пухлини призводять до обструкції ампули ДПК, що супроводжується жовтяницею або панкреатитом. Соматостатиноми дуже рідко супроводжуються функціональними синдромами [90].
Дуоденальні гангліоцитарні парагангліоми також переважно розташовані у періампулярній зоні. Вони часто великих розмірів (>2 см) та поширюються у м’язовий шар. Проте, на відміну від гастриноми або соматостатиноми, ці пухлини мають доброякісний перебіг [85, 90].
Низькодиференційовані ДПК-НЕП найчастіше виявляють у ділянці фатерового сосочка або у періампулярній зоні. Це гормонально неактивні пухлини і зазвичай на момент діагностики первинної пухлини у пацієнтів виявляють Мтс у ЛВ або печінці [90].
НЕП у ділянці фатерового сосочка прогностично відрізняються від таких у періампулярній зоні. Перші найчастіше виявляють у термінальній стадії, вони мають гірший прогноз, ніж ДПК-НЕП інших локалізацій [77]. Розмір ДПК-НЕП ампулярної зони не корелює з Мтс у печінці, на відміну від ДПК-НЕП інших локалізацій [3].
Особливості ендоскопічної діагностики ДПК-НЕП. ДПК-НЕП, як і Ш-НЕП, виникають глибоко у слизовій оболонці та поширюються на підслизовий шар. Внаслідок цього ДПК-НЕП мають напівсферичну форму з рівномірним підвищенням, з крутим краєм, на відміну від менш вираженого при інших субмукозних пухлинах. Поверхня пухлини має колір нормальної слизової оболонки з жовтуватим відтінком. При збільшенні пухлини у розмірах у центрі формується втягнення слизової оболонки, яке в подальшому стає кратероподібним із виразкуванням (рис. 2).
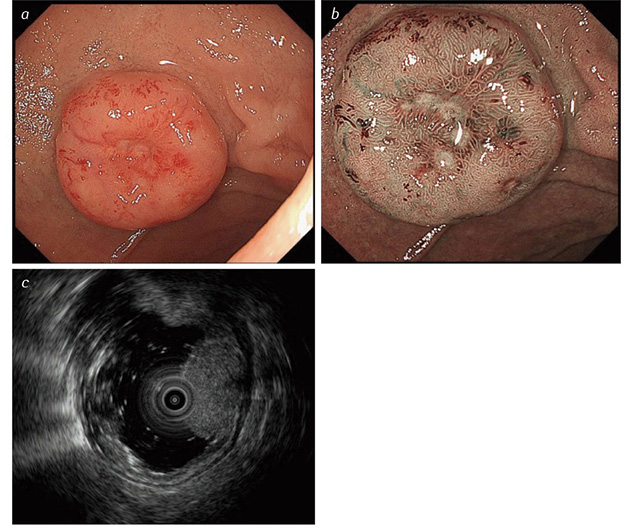
Диференційну діагностику ДПК-НЕП слід проводити з гіперплазією брунерівських залоз, аденомою, аденокарциномою, GIST, лімфоїдною гіперплазією, Мтс, нейрофібромою та шванномою [91].
ЕГДС та біопсія пухлини є найбільш ефективними методами діагностики ДПК-НЕП. Але не завжди вдається за допомогою біопсії отримати пухлинний матеріал через глибоке розташування пухлини у підслизовому шарі.
Ендоскопічне УЗД має важливе значення для підтвердження розміру пухлини та глибини інвазії. КТ та МРТ призначаються у випадку підозри або доведеної поширеної хвороби, наявності Мтс.
Лікування ДПК-НЕП. Методи лікування пацієнтів із ДПК-НЕП залежать від розміру, локалізації, ступеня градації G, стадії та типу пухлини. Але сьогодні не існує єдиного консенсусу щодо найбільш ефективних методів лікування при ДПК-НЕП.
ЕР може бути проведена у випадку невеликих за розміром функціонально неактивних G1 ДПК-НЕП (<10 мм), які розташовані у періампулярній зоні ділянки фатерового сосочка без Мтс. Якщо G1 ДПК-НЕП розміщені в ампулярній зоні великого дуоденального сосочка, тоді необхідно виконувати локальну резекцію з лімфаденектомією.
Методи лікування G2 ДПК-НЕП є суперечливими, а саме обмежена резекція у випадку великих НЕП >20 мм або НЕП будь-якого розміру з наявністю Мтс у ЛВ. ЕР можлива при високодиференційованих НЕП будь-якого розміру. Трансдуоденальна локальна резекція або панкреатодуоденальна резекція також застосовуються у лікуванні G2–3 ДПК-НЕП [92–94].
ЕР є безпечним та ефективним малоінвазивним методом лікування при ДПК-НЕП. Відомо декілька методик ЕР: видалення слизової кишки та субмукозна резекція стінки з використанням технік «endoloop and endoclip» або «over-the-score clip», ЕР всіх шарів стінки з лапароскопічним асистуванням [6, 99, 100]. Субмукозні резекції ДПК-НЕП нерідко супроводжуються кровотечею, особливо при розміщенні пухлини у дистальних відділах органа [97].
Хірургічна резекція рекомендована для пацієнтів зі спорадичними дуоденальними гастриномами [90], оскільки у половині випадків у них виявляють Мтс у ЛВ [73, 104, 105]. Також відомо, що резекція знижує ризик печінкових Мтс [82, 106]. Агресивний хірургічний підхід певною мірою виправданий у разі ДПК-НЕП, асоційованих з МЕН I типу та СЗЕ, з його застосуванням збільшується медіана загальної виживаності та знижується частота виникнення Мтс у печінці [69, 96].
Деякі автори пропонують провести дуоденектомію зі збереженням підшлункової залози у випадку дуоденальної гастриноми, що покращує віддалені результати [111, 112].
Хворих на ДПК-НЕП, локалізовані в ділянці ампули або у періампулярній зоні великого дуоденального сосочка, слід лікувати шляхом резекції стінки кишки або ендоскопічної папілектомії [17, 113].
Спостереження. ЕГДС, УЗД черевної порожнини/КТ, визначення рівня хромограніну А проводяться на 6; 24-му і 36-му місяці після лікування з приводу ДПК-НЕП.
Після резекції стінки ДПК спостереження проводиться протягом як мінімум 3 років і складається з КТ, соматостатин-рецепторної сцинтиграфії та визначення рівня хромограніну А на 6-й і 12-й місяць, згодом — щорічно протягом 3 років.
Оцінка стану пацієнтів, у яких виявлено нерезектабельну ДПК-НЕП та Мтс, має бути проведена з використанням КТ, соматостатин-рецепторної сцинтиграфії з інтервалом кожні 3 та 6 міс [3], визначенням хромограніну А.
Рівень хромограніну асоціюється с прогресуванням захворювання, тому може визначатися постійно з певними інтервалами до встановлення можливого подальшого розвитку хвороби.
Принципи лікування пацієнтів з нерезектабельними або метастатичними Ш-НЕП та ДПК-НЕП [114]. Ключові пункти:
- системна терапія може бути недоцільна у рутинному застосуванні, проводить індивідуально у кожному конкретному випадку та потребує мультидисциплінарного підходу для вибору оптимального способу лікування, а саме: спостереження за пацієнтами з нерезектабельними НЕП, регіонарна внутрішньопечінкова терапія у випадку, коли тільки наявні Мтс у печінці, циторедуктивна хірургія, системна терапія;
- наразі немає жодних рекомендацій, які б запропонували чітку послідовність застосування вищевказаних методів лікування хворих із поширеними Ш-НЕП та ДПК-НЕП;
- не визначена роль ад’ювантної системної терапії після хірургічного лікування з приводу Ш-НЕП та ДПК-НЕП.
Згідно з сучасними стандартами лікування пацієнтів із нерезектабельними та метастатичними НЕП ШКТ запропоновано схеми та препарати, наведені у табл. 5.
Таблиця 5. Варіанти системного лікування пацієнтів із нерезектабельними та метастатичними Ш-НЕП та ДПК-НЕП (NCCN 3.2017)*
| Нерезектабельні Ш-НЕП та ДПК-НЕП | 1) G1–2:
2) G3/прогресування:
3) Пептидна рецепторна цільова терапія (PRRT) з використанням аналогів октреотиду з Лютецієм-177 4) MIBG — внутрішня радіотерапія з метайодбензилгуанідином |
| Карциноїдний синдром | Ланреотид 120 мг кожні 28 діб Октреотид LAR 2–30 мг кожні 28 діб Звичайний октреотид 200 мг 3 рази на добу щодня |
*Аналіз методів лікування з оглядом літератури описаний у розділі, присвяченому НЕП тонкої кишки.
Список використаної та рекомендованої літератури
- Lawrence B., Gustafsson B.I., Chan A. et al. (2011) The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol. Metab. Clin. North Am., 40: 1–18, vii [PMID: 21349409 DOI: 10.1016/j.ecl.2010.12.005].
- Yao J.C., Hassan M., Phan A. et al. (2008) One hundred years after «carcinoid»: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol., 26: 3063–3072 [PMID: 18565894 DOI: 10.1200/JCO.2007.15.4377].
- Delle Fave G., Kwekkeboom D.J., Van Cutsem E. et al. (2012) ENETS consensus guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology, 95: 74–87.
- O’Connor J.M., Marmissolle F., Bestani C. et al. (2014) Observational study of patients with gastro-enteropancreatic and bronchial neuroendocrine tumors in Argentina: results from the large database of a multidisciplinary group clinical multicenter study. Mol. Clin. Oncol., 2: 673–684.
- Modlin I.M., Lye K.D., Kidd M. (2004) A 50-year analysis of 562 gastric carcinoids: small tumor or larger problem? Am. J. Gastroenterol., 99: 23–32.
- Ballian N., Loeffler A.G., Rajamanickam V. et al. (2009) A simplified prognostic system for resected pancreatic neuroendocrine neoplasms. HPB (Oxford),11: 422–428.
- Klimstra D.S., Modlin I.R., Adsay N.V. et al. (2010) Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am. J. Surg. Pathol., 34: 300–313.
- Qian Z.R., Ter-Minassian M., Chan J.A. et al. (2013) Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. J. Clin. Oncol., 31: 3418–3425.
- Francis J.M., Kiezun A., Ramos A.H. et al. (2013) Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat. Genet., 45: 1483–1486.
- Kim H.S., Lee H.S., Nam K.H. et al. (2014) p27 loss is associated with poor prognosis in gastroenteropancreatic neuroendocrine tumors. Cancer Res. Treat., 46: 383–392.
- Khan M.S., Kirkwood A., Tsigani T. et al. (2013) Circulating tumor cells as prognostic markers in neuroendocrine tumors. J. Clin. Oncol., 31: 365–372.
- Mori H., Shintaro F., Kobara H. et al. (2013) Successful closing of duodenal ulcer after endoscopic submucosal dissection with over-the-scope clip to prevent delayed perforation. Dig. Endosc., 25: 459–461 [PMID: 23368742 DOI: 10.1111/j.1443-1661.2012.01363. x].
- Ellis L., Shale M.J., Coleman M.P. (2010) Carcinoid tumors of the gastrointestinal tract: trends in incidence in England since 1971. Am. J. Gastroenterol., 105: 2563–2569 [PMID: 20823835 DOI: 10.1038/ajg.2010.341].
- Ito T., Sasano H., Tanaka M. et al. (2010) Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J. Gastroenterol., 45: 234–243 [PMID: 20058030 DOI: 10.1007/ s00535-009-0194-8].
- Niederle M.B., Hackl M., Kaserer K., Niederle B. (2010) Gastroentero-pancreatic neuroendocrine tumours: the current incidence and staging based on the WHO and European Neuroendocrine Tumour Society classi cation: an analysis based on prospectively collected parameters. Endocr. Relat. Cancer., 17: 909–918 [PMID: 20702725 DOI: 10.1677/ERC-10-0152].
- Cho M.Y., Kim J.M., Sohn J.H. et al. (2012) Current Trends of the Incidence and Pathological Diagnosis of Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs) in Korea 2000–2009: Multicenter Study. Cancer Res. Treat., 44: 157–165 [PMID: 23091441 DOI: 10.4143/crt.2012.44.3.157].
- Caldarella A., Crocetti E., Paci E. (2011) Distribution, incidence, and prognosis in neuroendocrine tumors: a population based study from a cancer registry. Pathol. Oncol. Res., 17: 759–763 [PMID: 21476126 DOI: 10.1007/s12253-011-9382-y].
- Boyce M., Thomsen L. (2015) Gastric neuroendocrine tumors: prevalence in Europe, USA, and Japan, and rationale for treatment with a gastrin/CCK2 receptor antagonist. Scand. J. Gastroenterol., 50: 550–559 [PMID: 25665655 DOI: 10.3109/00365521.2015.100994 1].
- Ito T., Igarashi H., Nakamura K. et al. (2015) Epidemiological trends of pancreatic and gastrointestinal neuroendocrine tumors in Japan: a nationwide survey analysis. J. Gastroenterol., 50: 58–64 [PMID: 24499825 DOI: 10.1007/s00535-014-0934-2].
- Nikou G.C., Angelopoulos T.P. (2012) Current concepts on gastric carcinoid tumors. Gastroenterol. Res. Pract., 2012: 287825 [PMID: 23316222 DOI: 10.1155/2012/287825].
- Basuroy R., Srirajaskanthan R., Prachalias A. et al. (2014) Review article: the investigation and management of gastric neuroendocrine tumours. Aliment Pharmacol. Ther., 39: 1071–1084 [PMID: 24628514 DOI: 10.1111/apt.12698].
- Li T.T., Qiu F., Qian Z.R. et al. (2014) Classification, clinicopathologic features and treatment of gastric neuroendocrine tumors. World J. Gastroenterol., 20: 118–125 [PMID: 24415864 DOI: 10.3748/wjg.v20.i1.118].
- Massironi S., Sciola V., Spampatti M.P. et al. (2009)Gastric carcinoids: between underestimation and overtreatment. World J. Gastroenterol., 15: 2177–2183 [PMID: 19437556 DOI: 10.3748/wjg.15.2177].
- Zhang L., Ozao J., Warner R., Divino C. (2011) Review of the pathogenesis, diagnosis, and management of type I gastric carcinoid tumor. World J. Surg., 35: 1879–1886 [PMID: 21559999 DOI: 10.1007/s00268-011-1137-0].
- Scherübl H., Cadiot G., Jensen R.T. et al. (2010) Neuroendocrine tumors of the stomach (gastric carcinoids) are on the rise: small tumors, small problems? Endoscopy, 42: 664–671 [PMID: 20669078 DOI: 10.1055/s-0030-1255564].
- Kaltsas G., Grozinsky-Glasberg S., Alexandraki K.I. et al. (2014) Current concepts in the diagnosis and management of type 1 gastric neuroendocrine neoplasms. Clin. Endocrinol. (Oxf), 81: 157–168 [PMID: 24750249 DOI: 10.1111/cen.12476].
- O’Toole D., Delle Fave G., Jensen R.T. (2012) Gastric and duodenal neuroendocrine tumours. Best Pract. Res. Clin. Gastroenterol., 26: 719–735 [PMID: 23582915 DOI: 10.1016/j.bpg.2013.01.002].
- Dakin G.F., Warner R.R., Pomp A. et al. (2006) Presentation, treatment, and outcome of type 1 gastric carcinoid tumors. J. Surg. Oncol., 93: 368–372 [PMID: 16550587 DOI: 10.1002/jso.20468].
- Sato Y., Iwafuchi M., Ueki J. et al. (2002) Gastric carcinoid tumors without autoimmune gastritis in Japan: a relationship with Helicobacter pylori infection. Dig. Dis. Sci., 47: 579–585 [PMID: 11911346].
- Sato Y. (2015) Endoscopic diagnosis and management of type I neuroendocrine tumors. World J. Gastrointest. Endosc., 7: 346–353 [PMID: 25901213 DOI: 10.4253/wjge.v7.i4.346].
- Sato Y., Imamura H., Kaizaki Y. et al. (2014) Management and clinical outcomes of type I gastric carcinoid patients: retrospective, multicenter study in Japan. Dig. Endosc., 26: 377–384 [PMID: 24188531 DOI: 10.1111/den.12197].
- Meko J.B., Norton J.A. (1995) Management of patients with Zollinger-Ellison syndrome. Annu. Rev. Med., 46: 395–411 [PMID: 7598474].
- Nikou G.C., Toubanakis C., Nikolaou P. et al. (2005)Gastrinomas associated with MEN-1 syndrome: new insights for the diagnosis and management in a series of 11 patients. Hepatogastroenterology, 52: 1668–1676 [PMID: 16334754].
- Kim G.H., Kim J.I., Jeon S.W. et al. (2014) Endoscopic resection for duodenal carcinoid tumors: a multicenter, retrospective study. J. Gastroenterol. Hepatol., 29: 318–324.
- Crosby D.A., Donohoe C.L., Fitzgerald L. et al. (2012) Gastric neuroendocrine tumours. Dig. Surg., 29: 331–348 [PMID: 23075625 DOI: 10.1159/000342988].
- Singh R., Yao K., Anagnostopoulos G. et al. (2008) Microcarcinoid tumor diagnosed with high-resolution magni cation endoscopy and narrow band imaging. Endoscopy, 40 (Suppl 2): E12 [PMID: 18278715 DOI: 10.1055/s-2007-995393].
- Karaca C., Turner B.G., Cizginer S. et al. (2010) Accuracy of EUS in the evaluation of small gastric subepithelial lesions. Gastrointest. Endosc., 71: 722–727 [PMID: 20171632 DOI: 10.1016/j.gie.2009.10.019].
- Bushnell D.L., Baum R.P. (2011) Standard imaging techniques for neuroendocrine tumors. Endocrinol. Metab. Clin. North Am., 40: 153–162, ix [PMID: 21349416 DOI: 10.1016/j.ecl.2010.12.002].
- Sato Y., Hashimoto S., Mizuno K. et al. (2016) Management of gastric and duodenal neuroendocrine tumors. World J. Gastroenterol., 22(30): 6817–6828.
- Kjaer A., Knigge U. (2015) Use of radioactive substances in diagnosis and treatment of neuroendocrine tumors. Scand J. Gastroenterol., 50: 740–747 [PMID: 25959100 DOI: 10.3109/00365521.2015.103 3454].
- Johnbeck C.B., Knigge U., Kjær A. (2014) PET tracers for somatostatin receptor imaging of neuroendocrine tumors: current status and review of the literature. Future Oncol., 10: 2259–2277 [PMID: 25471038 DOI: 10.2217/fon.14.139].
- Kulke M.H., Shah M.H., Benson A.B. et al. (2015) Neuroendocrine tumors, version 1.2015. J. Natl. Compr. Cancer Netw., 13: 78–108 [PMID: 25583772].
- Sato Y., Takeuchi M., Hashimoto S. et al. (2013) Usefulness of endoscopic submucosal dissection for type I gastric carcinoid tumors compared with endoscopic mucosal resection. Hepatogastroenterology, 60: 1524–1529 [PMID: 23933946 DOI: 10.5754/hge121185].
- Kim H.H., Kim G.H., Kim J.H. et al. (2014) The efficacy of endoscopic submucosal dissection of type I gastric carcinoid tumors compared with conventional endoscopic mucosal resection. Gastroenterol Res. Pract., 2014: 253860 [PMID: 24693280 DOI: 10.1155/2014/253860].
- Ravizza D., Fiori G., Trovato C. et al. (2007) Long-term endoscopic and clinical follow-up of untreated type 1 gastric neuroendocrine tumours. Dig. Liver Dis., 39: 537–543 [PMID: 17433795 DOI: 10.1016/j.dld.2007.01.018].
- Campana D., Ravizza D., Ferolla P. et al. (2016) Clinical management of patients with gastric neuroendocrine neoplasms associated with chronic atrophic gastritis: a retrospective, multicentre study. Endocrine, 51: 131–139 [PMID: 25814125 DOI: 10.1007/s12020-015-0584-z].
- Gladdy R.A., Strong V.E., Coit D. et al. (2009) De ning surgical indications for type I gastric carcinoid tumor. Ann. Surg. Oncol., 16: 3154–3160 [PMID: 19727959 DOI: 10.1245/s10434-009-0687-y].
- Jenny H.E., Ogando P.A., Fujitani K. et al. (2016) Laparoscopic antrectomy: a safe and definitive treatment in managing type 1 gastric carcinoids. Am. J. Surg., 211: 778–782 [PMID: 26992358 DOI: 10.1016/j.amjsurg.2015.08.040].
- Ozao-Choy J., Buch K., Strauchen J.A. et al. (2010) Laparoscopic antrectomy for the treatment of type I gastric carcinoid tumors. J. Surg. Res., 162: 22–25 [PMID: 20421108 DOI: 10.1016/j.jss.2010.01.005].
- Thomas D., Tsolakis A.V., Grozinsky-Glasberg S. et al. (2013) Long-term follow-up of a large series of patients with type 1 gastric carcinoid tumors: data from a multicenter study. Eur. J. Endocrinol., 168: 185–193 [PMID: 23132699 DOI: 10.1530/EJE-12-0836].
- Grozinsky-Glasberg S., Kaltsas G., Gur C. et al. (2008) Long-acting somatostatin analogues are an effective treatment for type 1 gastric carcinoid tumours. Eur. J. Endocrinol., 159: 475–482 [PMID: 18662970 DOI: 10.1530/EJE-08-0420].
- Campana D., Nori F., Pezzilli R. et al. (2008) Gastric endocrine tumors type I: treatment with long-acting somatostatin analogs. Endocr. Relat. Cancer, 15: 337–342 [PMID: 18310299 DOI: 10.1677/ ERC-07-0251].
- Manfredi S., Pagenault M., de Lajarte-Thirouard A.S., Bretagne J.F. (2007) Type 1 and 2 gastric carcinoid tumors: long-term follow-up of the efficacy of treatment with a slow-release somatostatin analogue. Eur. J. Gastroenterol. Hepatol., 19: 1021–1025 [PMID: 18049175 DOI: 10.1097/MEG.0b013e328220eae0].
- Jianu C.S., Fossmark R., Syversen U. et al. (2011) Five-year follow-up of patients treated for 1 year with octreotide long-acting release for enterochromaffin-like cell carcinoids. Scand. J. Gastroenterol., 46: 456–463 [PMID: 21133821 DOI: 10.3109/00365521.2010.539255].
- Massironi S., Zilli A., Conte D. (2015) Somatostatin analogs for gastric carcinoids: For many, but not all. World J. Gastroenterol., 21: 6785–6793 [PMID: 26078554 DOI: 10.3748/wjg.v21.i22.6785].
- Massironi S., Zilli A., Fanetti I. et al. (2015) Intermittent treatment of recurrent type-1 gastric carcinoids with somatostatin analogues in patients with chronic autoimmune atrophic gastritis. Di. Liver Dis., 47: 978–983 [PMID: 26321479 DOI: 10.1016/j.dld.2015.07.155].
- Fossmark R., Sørdal Ø., Jianu C.S. et al. (2012) Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment Pharmacol. Ther., 36: 1067–1075 [PMID: 23072686 DOI: 10.1111/apt.12090].
- Moore A.R., Boyce M., Steele I.A. et al. (2013) Netazepide, a gastrin receptor antagonist, normalises tumour biomarkers and causes regression of type 1 gastric neuroendocrine tumours in a nonrandomised trial of patients with chronic atrophic gastritis. PLoS One, 8: e76462 [PMID: 24098507 DOI: 10.1371/journal.pone.0076462].
- La Rosa S., Vanoli A. (2014) Gastric neuroendocrine neoplasms and related precursor lesions. J. Clin. Pathol., 67: 938–948 [PMID: 25053544 DOI: 10.1136/jclinpath-2014-202515].
- Lahner E., Pilozzi E., Esposito G. et al. (2014) Gastric carcinoid in the absence of atrophic body gastritis and with low Ki67 index: a clinical challenge. Scand. J. Gastroenterol., 49: 506–510.
- La Rosa S., Inzani F., Vanoli A. et al. (2011) Histologic characterization and improved prognostic evaluation of 209 gastric neuroendocrine neoplasms. Hum. Pathol., 42: 1373–1384.
- Rindi G., Azzoni C., La Rosa S. et al. (1999) ECL cell tumor and poorly differentiated endocrine carcinoma of the stomach: prognostic evaluation by pathological analysis. Gastroenterology, 116: 532–542.
- Cavallaro A., Zanghì A., Cavallaro M. et al. (2014) The role of 68-Ga-DOTATOC CT-PET in surgical tactic for gastric neuroendocrine tumors treatment: our experience: a case report. Int. J. Surg., 12(suppl. 1): S225–S231.
- Alexander H.R., Fraker D.L., Norton J.A. et al. (1998) Prospective study of somatostatin receptor scintigraphy and its effect on operative outcome in patients with Zollinger-Ellison syndrome. Ann. Surg., 228: 228–238.
- Merola E., Sbrozzi-Vanni A., Panzuto F. et al. (2012) Type I gastric carcinoids: a prospective study on endoscopic management and recurrence rate. Neuroendocrinology, 95: 207–213.
- Uygun A., Kadayifci A., Polat Z. et al. (2014) Long- term results of endoscopic resection for type I gastric neuroendocrine tumors. J. Surg. Oncol.,109: 71–74.
- Grozinsky-Glasberg S., Thomas D., Strosberg J.R. et al. (2013) Metastatic type 1 gastric carcinoid: a real threat or just a myth? World J. Gastroenterol., 19: 8687–8695.
- Chen W.F., Zhou P.H., Li Q.L. et al. (2012) Clinical impact of endoscopic submucosal dissection for gastric neuroendocrine tumors: a retrospective study from mainland China. Sci. World J., 2012: 869769.
- Kobara H., Mori H., Rafiq K. et al. (2013) Indications of endoscopic submucosal dissection for symptomatic benign gastrointestinal subepithelial or carcinoid tumors originating in the submucosa. Mol. Clin. Oncol., 1: 1002–1008.
- Kwon Y.H., Jeon S.W., Kim G.H et al. (2013) Long- term follow up of endoscopic resection for type 3 gastric NET. World J. Gastroenterol., 19: 8703–8708.
- Yao J.C., Hassan M., Phan A. et al. (2008) One Hundred Years After «Carcinoid»: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. Clin. Oncol., 26: 3063–3072.
- Modlin I.M., Champaneria M.C., Chan A.K., Kidd M. (2007) A three-decade analysis of 3,911 small intestinal neuroendocrine tumors: the rapid pace of no progress. Am. J. Gastroenterol., 102: 1464–1473 [PMID: 17391319 DOI: 10.1111/j.1572-0241.2007.01185.x].
- Hoffmann K.M., Furukawa M., Jensen R.T. (2005) Duodenal neuroendocrine tumors: Classification, functional syndromes, diagnosis and medical treatment. Best Pract. Res. Clin. Gastroenterol., 19: 675–697 [PMID: 16253893 DOI: 10.1016/ j.bpg.2005.05.009].
- Soga J. (2003) Endocrinocarcinomas (carcinoids and their variants) of the duodenum. An evaluation of 927 cases. J. Exp. Clin. Cancer Res., 22: 349–363 [PMID: 14582691].
- Burke A.P., Sobin L.H., Federspiel B.H. et al. (1990) Carcinoid tumors of the duodenum. A clinicopathologic study of 99 cases. Arch. Pathol. Lab. Med., 114: 700–704 [PMID: 1694655].
- Klöppel G., Arnold R., Capella C. et al. (2010) Neuroendocrine neoplasms of the ampullary resion. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO classication of Tumours of the digestive system. Lyon: IARC: 13–14.
- Randle R.W., Ahmed S., Newman N.A., Clark C.J. (2014) Clinical outcomes for neuroendocrine tumors of the duodenum and ampulla of Vater: a population-based study. J. Gastrointest Surg., 18: 354–362 [PMID: 24114680 DOI: 10.1007/s11605-013-2365-4].
- Jensen R.T., Berna M.J., Bingham D.B., Norton J.A. (2008) Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer, 113: 1807–1843 [PMID: 18798544 DOI: 10.1002/cncr.23648].
- Gibril F., Schumann M., Pace A., Jensen R.T. (2004) Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore), 83: 43–83 [PMID: 14747767 DOI: 10.1097/01.md.0000112297.72510.32].
- Levy A.D., Taylor L.D., Abbott R.M., Sobin L.H. (2005) Duodenal carcinoids: imaging features with clinical-pathologic comparison. Radiology, 237: 967–972 [PMID: 16237144 DOI: 10.1148/ radiol.2373041863].
- Weber H.C., Venzon D.J., Lin J.T. et al. (1995) Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology, 108: 1637–1649 [PMID: 7768367 DOI: 10.1016/0016-5085(95)90 124-8].
- Yu F., Venzon D.J., Serrano J. et al. (1999) Prospective study of the clinical course, prognostic factors, causes of death, and survival in patients with long-standing Zollinger-Ellison syndrome. J. Clin. Oncol., 17: 615–630 [PMID: 10080607].
- Chang S., Choi D., Lee S.J. et al. (2007) Neuroendocrine neoplasms of the gastrointestinal tract: classification, pathologic basis, and imaging features. Radiographics, 27: 1667–1679 [PMID: 18025510 DOI: 10.1148/rg.276075001].
- Kirshbom P.M., Kherani A.R., Onaitis M.W. et al. (1999) Foregut carcinoids: a clinical and biochemical analysis. Surgery, 126: 1105–1110 [PMID: 10598194 DOI: 10.1067/msy.2099.101430].
- Klöppel G. (2007) Tumour biology and histopathology of neuroendocrine tumours. Best Pract. Res. Clin. Endocrinol. Metab., 21: 15–31 [PMID: 17382263 DOI: 10.1016/j.beem.2007.01.004].
- Grin A., Kim Y.I., Mustard R. et al. (2012) Duodenal gastrinoma with multiple gastric neuroendocrine tumors secondary to chronic Helicobacter pylori gastritis. Am. J. Surg. Pathol., 36: 935–940 [PMID: 22588069 DOI: 10.1097/PAS.0b013e31824babc2].
- Merchant S.H., Vander Jagt T., Lathrop S., Amin M.B. (2006) Sporadic duodenal bulb gastrin-cell tumors: association with Helicobacter pylori gastritis and long-term use of proton pump inhibitors. Am. J. Surg. Pathol., 30: 1581–1587 [PMID: 17122515 DOI: 10.1097/01.pas.0000213326.86992.98].
- Levy A.D., Sobin L.H. (2007) From the archives of the AFIP: Gastrointestinal carcinoids: imaging features with clinicopathologic comparison. Radiographics, 27: 237–257 [PMID: 17235010 DOI: 10.1148/rg.271065169]
- Thompson J.C., Lewis B.G., Wiener I., Townsend C.M. (1983) The role of surgery in the Zollinger-Ellison syndrome. Ann. Surg., 197: 594–607 [PMID: 6847279 DOI: 10.1097/00000658-198305000-00 014].
- Scherübl H., Jensen R.T., Cadiot G. et al. (2010) Neuroendocrine tumors of the small bowels are on the rise: Early aspects and management. World J. Gastrointest. Endosc., 2: 325–334 [PMID: 21160582 DOI: 10.4253/wjge.v2.i10.325].
- Waisberg J., Joppert-Netto G., Vasconcellos C. et al. (2013) Carcinoid tumor of the duodenum: a rare tumor at an unusual site. Case series from a single institution. Arq. Gastroenterol., 50: 3–9 [PMID: 23657299 DOI: 10.1590/ S0004-28032013000100002].
- Soga J. (2005) Early-stage carcinoids of the gastrointestinal tract: an analysis of 1914 reported cases. Cancer, 103: 1587–1595 [PMID: 15742328 DOI: 10.1002/cncr.20939].
- Mullen J.T., Wang H., Yao J.C. et al. (2005)
Carcinoid tumors of the duodenum. Surgery, 138:
971–977; discussion 977–978 [PMID: 16360380 DOI: 10.1016/j.surg.2005.09.016]. - Margonis G.A., Samaha M., Kim Y. et al. (2016) A multi-institutional analysis of duodenal neuroendocrine tumors: tumor biology rather than extent of resection dictates prognosis. J. Gastrointest. Surg., 20: 1098–1105 [PMID: 27008594 DOI: 10.1007/s11605-016-3135-x].
- Karagiannis S., Eshagzaiy K., Duecker C. et al. (2009) Endoscopic resection with the cap technique of a carcinoid tumor in the duodenal bulb. Endoscopy, 41(Suppl. 2): E288–E289 [PMID: 19899043 DOI: 10.1055/ s-0029-1215123].
- Matsumoto S., Miyatani H., Yoshida Y. (2015) Future directions of duodenal endoscopic submucosal dissection. World J. Gastrointest. Endosc., 7: 389–395 [PMID: 25901218 DOI: 10.4253/wjge. v7.i4.389].
- Hoteya S., Kaise M., Iizuka T. et al. (2015) Delayed bleeding after endoscopic submucosal dissection for non-ampullary superficial duodenal neoplasias might be prevented by prophylactic endoscopic closure: analysis of risk factors. Dig. Endosc., 27: 323–330 [PMID: 25186455 DOI: 10.1111/den.12377].
- Matsumoto S., Miyatani H., Yoshida Y., Nokubi M. (2011) Duodenal carcinoid tumors: 5 cases treated by endoscopic submucosal dissection. Gastrointest. Endosc., 74: 1152–1156 [PMID: 21944312 DOI: 10.1016/j.gie.2011.07.029].
- Takimoto K., Imai Y., Matsuyama K. (2014) Endoscopic tissue shielding method with polyglycolic acid sheets and fibrin glue to prevent delayed perforation after duodenal endoscopic submucosal dissection. Dig. Endosc., 26(Suppl. 2): 46–49 [PMID: 24750148 DOI: 10.1111/den.12280].
- Tsujimoto H., Ichikura T., Nagao S. et al. (2010) Minimally invasive surgery for resection of duodenal carcinoid tumors: endoscopic full-thickness resection under laparoscopic observation. Surg. Endosc., 24: 471–475 [PMID: 19517164 DOI: 10.1007/s00464-009-0574-4].
- Hari D.M., Goff S.L., Reich H.J. et al. (2013) Small bowel carcinoid: Location isn’t everything! World J. Gastrointest. Surg., 5: 239–244 [PMID: 23983905 DOI: 10.4240/wjgs.v5.i8.239].
- Jann H., Roll S., Couvelard A. et al. (2011) Neuroendocrine tumors of midgut and hindgut origin: tumor-node-metastasis classification determines clinical outcome. Cancer, 117: 3332–3341 [PMID: 21246527 DOI: 10.1002/cncr.25855].
- Jensen R.T., Niederle B., Mitry E.et al. (2006) Gastrinoma (duodenal and pancreatic). Neuroendocrinology, 84: 173–182 [PMID: 17312377 DOI: 10.1159/000098009].
- Norton J.A., Jensen R.T. (2004) Resolved and unresolved controversies in the surgical management of patients with Zollinger-Ellison syndrome. Ann. Surg., 240: 757–773 [PMID: 15492556 DOI: 10.1097/01.sla.0000143252.02142.3e].
- Norton J.A., Alexander H.R., Fraker D.L. et al. (2004) Does the use of routine duodenotomy (DUODX) affect rate of cure, development of liver metastases, or survival in patients with Zollinger-Ellison syndrome? Ann. Surg., 239: 617–625; discussion 626 [PMID: 15082965 DOI: 10.1097/01. sla.0000124290.05524.5e].
- Fraker D.L., Norton J.A., Alexander H.R. et al. (1994) Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann. Surg., 220: 320–328; discussion 328–330 [PMID: 7916560 DOI: 10.1097/00000658-199409000-00008].
- MacFarlane M.P., Fraker D.L., Alexander H.R. et al. (1995) Prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia type 1. Surgery, 118: 973–979; discussion 979–980 [PMID: 7491542 DOI: 10.1016/S0039-6060(05)80102-3].
- Jensen R.T., Cadiot G., Brandi M.L. et al. (2012) ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology, 95: 98–119 [PMID: 22261919 DOI: 10.1159/000335591].
- Norton J.A., Fraker D.L., Alexander H.R. et al. (2006) Surgery increases survival in patients with gastrinoma. Ann. Surg., 244: 410–419 [PMID: 16926567 DOI: 10.1097/01.sla.0000234802.44320.a5].
- Bartsch D.K., Fendrich V., Langer P. et al. (2005) Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann. Surg., 242: 757–764, discussion 764–766 [PMID: 16327485 DOI: 10.1097/01. sla.0000189549.51913.d8].
- Imamura M. (2010) Recent standardization of treatment strategy for pancreatic neuroendocrine tumors. World J. Gastroenterol., 16: 4519–4525 [PMID: 20857521 DOI: 10.3748/wjg.v16.i36.4519].
- Imamura M., Komoto I., Ota S. et al. (2011) Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J. Gastroenterol., 17: 1343–1353 [PMID: 21455335 DOI: 10.3748/wjg.v17.i10.1343].
- Makhlouf H.R., Burke A.P., Sobin L.H. (1999) Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer, 85: 1241–1249 [PMID: 10189128 DOI: 10.1002/(SI CI)1097-0142(19990315)85:6< 1241::AID-CNCR5>3.0.CO;2-4].
- NCCN Clinical Practice Guidelines in Oncology «Neuroendocrine Tumors» Version 3.2017 — June 13, 2017/NCCN.org.
Клинические рекомендации по диагностике и лечению нейроэндокринных опухолей желудка и двенадцатиперстной кишки
Национальный институт рака, Киев
Резюме. Нейроэндокринные опухоли (НЭО) объединяют довольно большую группу опухолей, характеризующихся биологическим разнообразием и гетерогенностью. По данным Всемирной организации здравоохранения, за последние годы отмечается повышение частоты выявления НЭО до 3,65 на 100 тис. населения в год. Стадия и дифференциация НЭО являются основными факторами прогноза у пациентов с НЭО желудочно-кишечного тракта (ЖКТ). Применение современных методов диагностики — компьютерной томографии, магнитно-резонансной томографии, сцинтиграфии, позитронно-эмиссионной томографии, объединенной с компьютерной томографией, и иммуногистохимического исследования биологического материала — играет ключевую роль в выявлении НЭО ЖКТ. Основные принципы хирургического лечения заключаются в адекватном объеме лимфодиссекции, удалении всех видимых первичных пальпабельных опухолей, устранении рецидива и метастазов. Медикаментозное лечение пациентов с НЭО ЖКТ IV стадии зависит от степени градации (G), индекса пролиферации (Ki-67) и митотической активности и заключается в применении пролонгированных октреотидов, иммунотерапии, системной химиотерапии и локорегионарной терапии. Медиана общей выживаемости пациентов с НЭО ЖКТ III стадии, получивших лечение, составляет 112–170 мес, III — 70–105 мес и IV — 13–60 мес. Рекомендации по диагностике и лечению пациентов с НЭО ЖКТ базируются на рекомендациях ENETS 2016 г., NCCN 03/2017, данные которых обновлялись на основе просмотра литературы на базе поискового ресурса PUBMED за 2015–2016 гг.
нейроэндокринные опухоли желудочно-кишечного тракта, клинические рекомендации, методы диагностики, хирургическое лечение, циторедуктивные операции, гормонотерапия, химиотерапия.
Адреса:
Зубарєв Микола Геннадійович
03022, Київ, вул. Ломоносова, 33/43
Національний інститут раку
E-mail: mykola.zubaryev@gmail.com





Leave a comment